
Understanding the Responses of Soil Bacterial Communities to Long-Term Fertilization Regimes Using DNA and RNA Sequencing


Abstract
:1. Introduction
2. Material and Methods
2.1. Experimental Design
2.2. Soil Sampling
2.3. Microbial DNA Extraction, RNA Isolation and cDNA Synthesis, PCR Amplification
2.4. 454-Pyrosequencing and Sequence Analysis
2.5. α- and β-Diversity Estimation, Taxonomy, and Statistical Analysis
2.6. Network Analysis
2.7. Metagenome Inference
3. Results
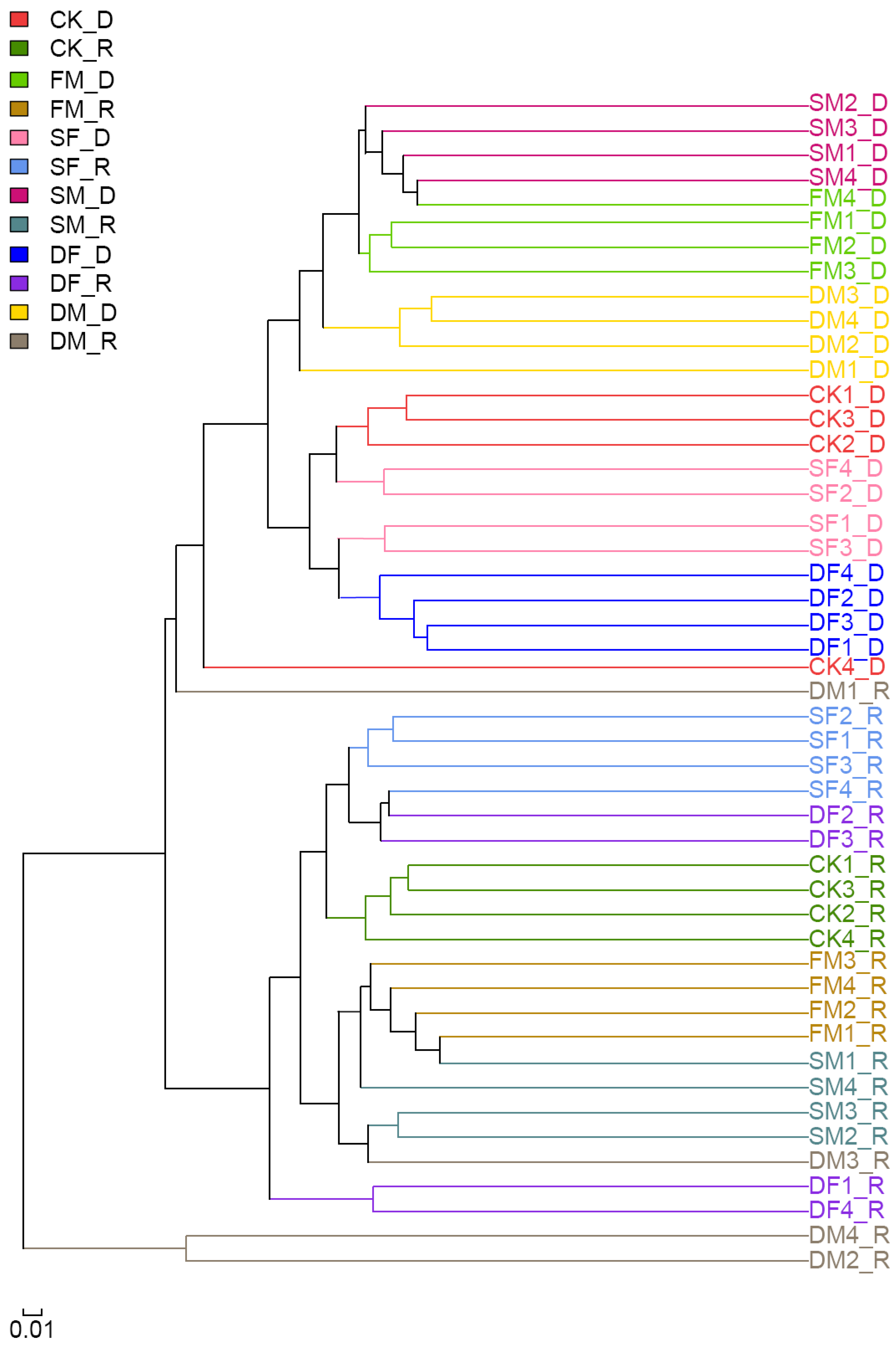
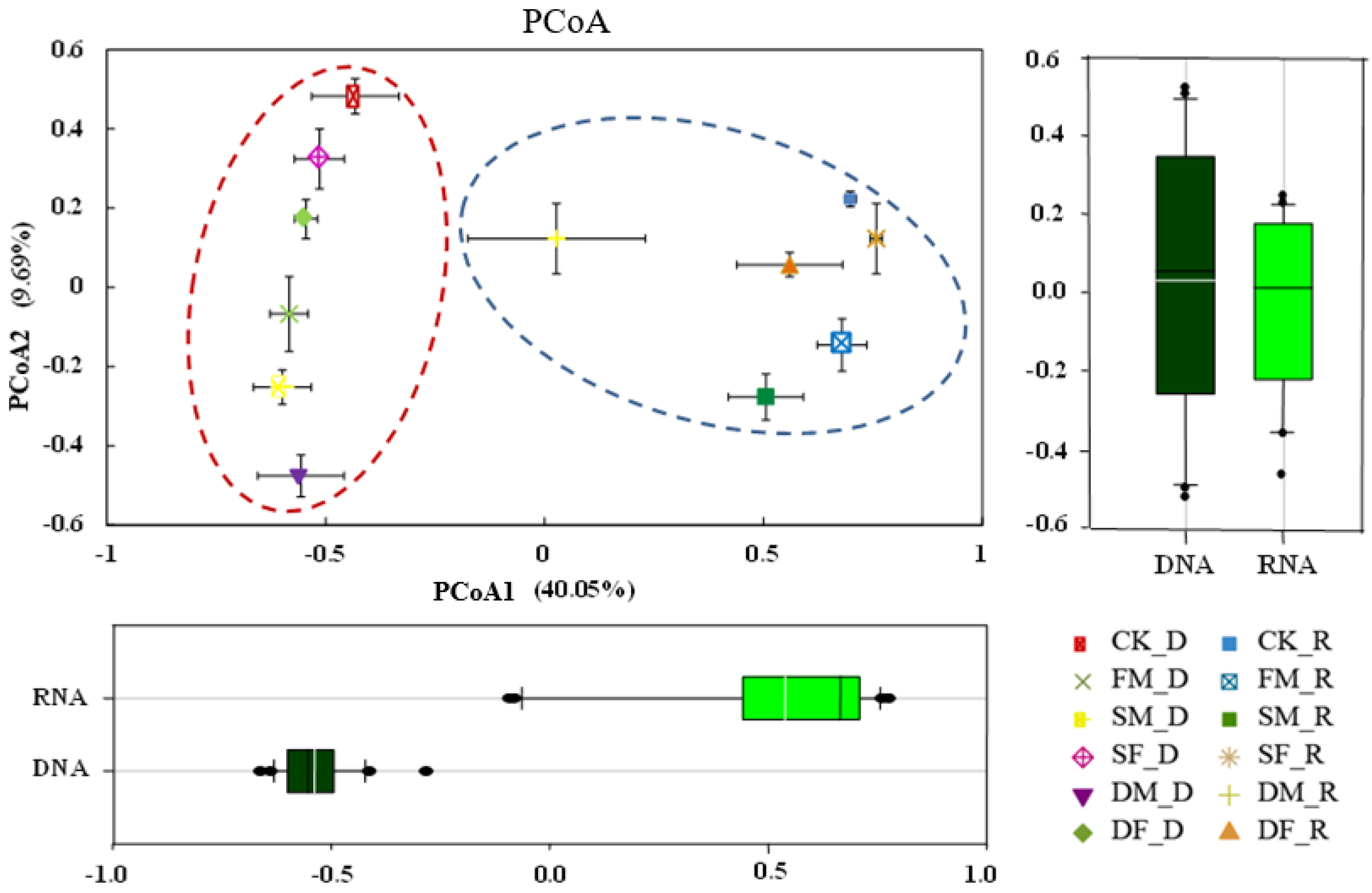
3.1. Quantity and Diversity of Soil Bacterial Community under Long-Term Fertilization Regimes
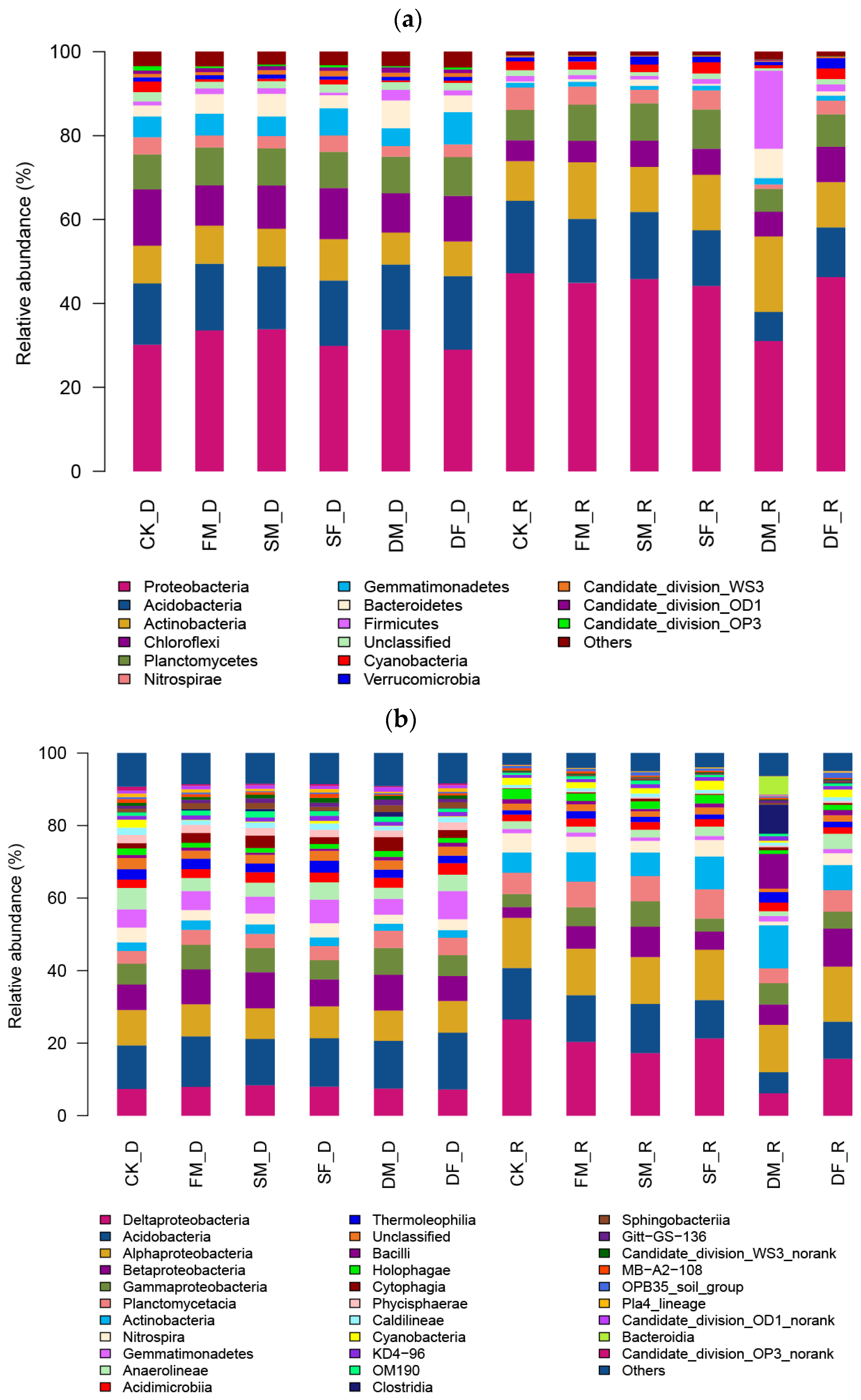
3.2. Structure and Composition of Soil Bacterial Taxa
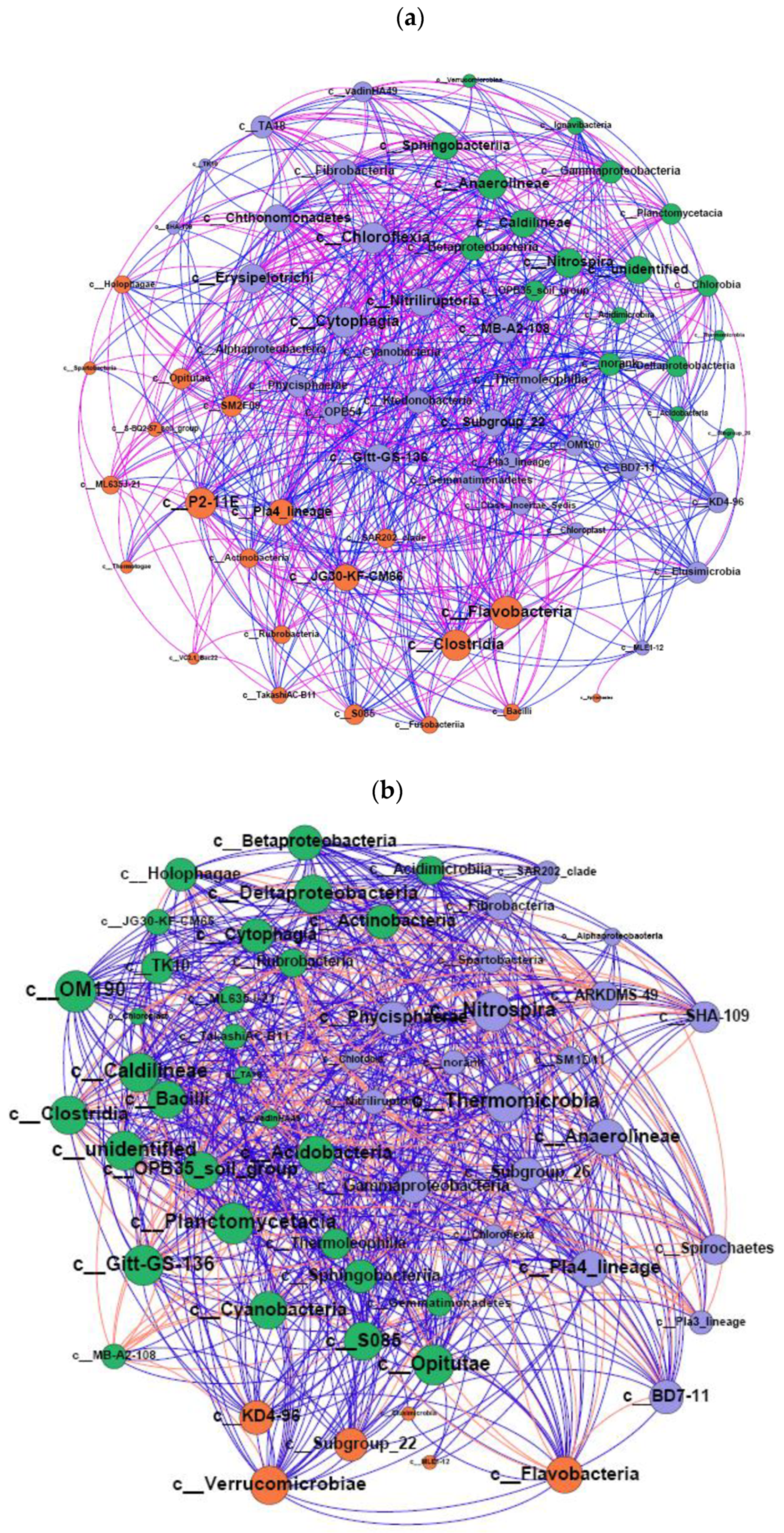
3.3. Network Analysis between DNA and RNA Levels
3.4. Functional Analysis between DNA and RNA Levels
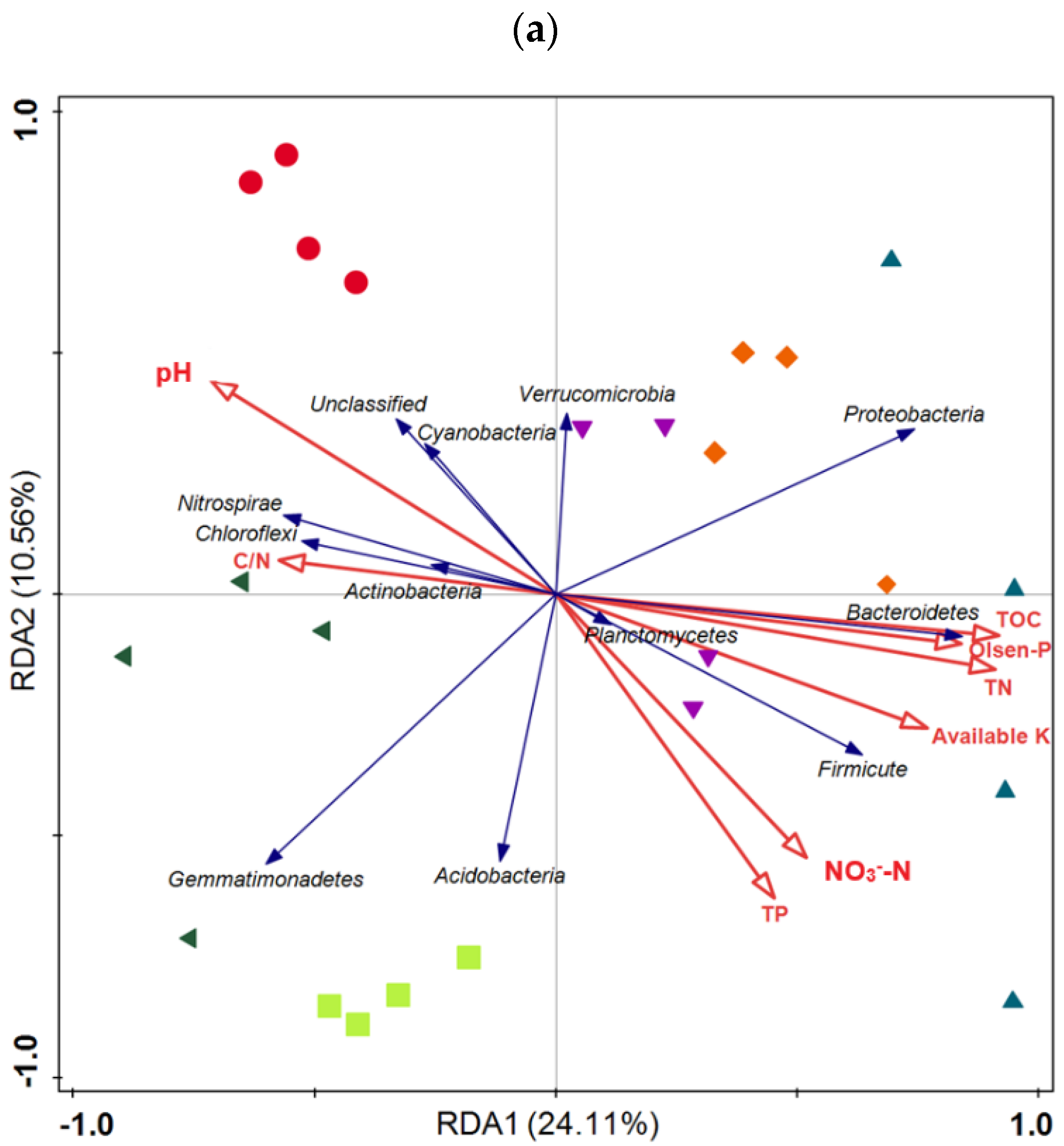
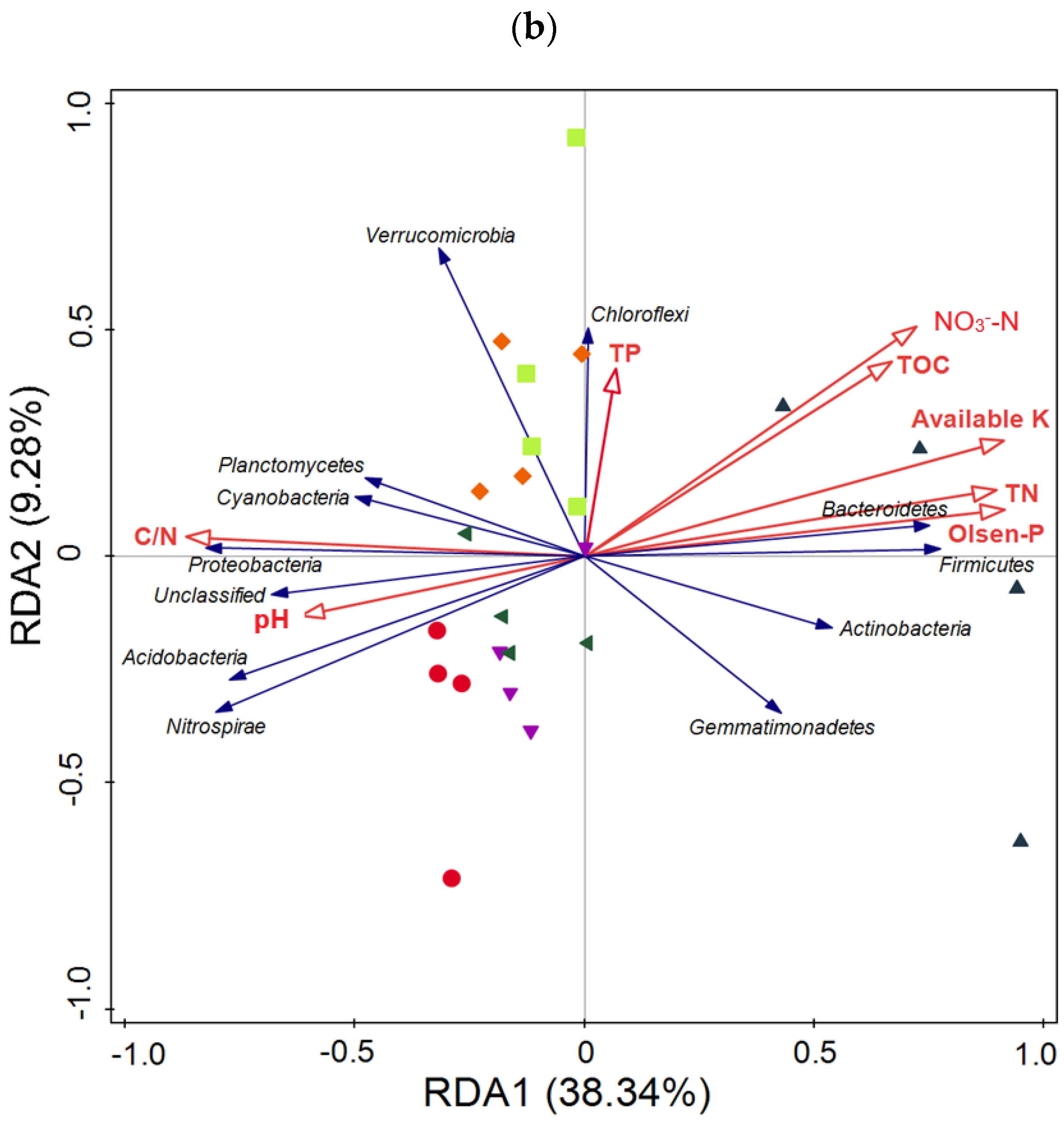
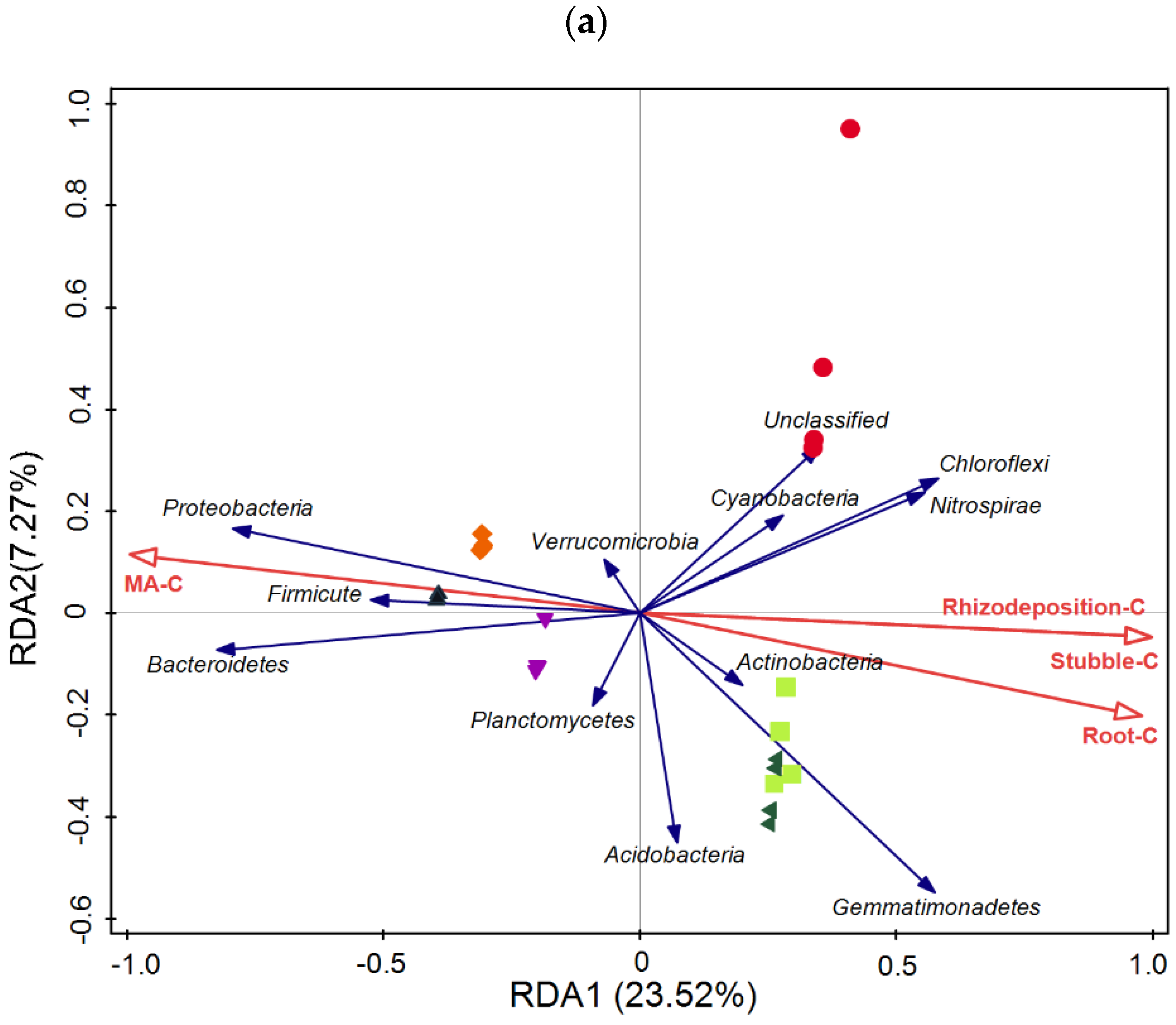
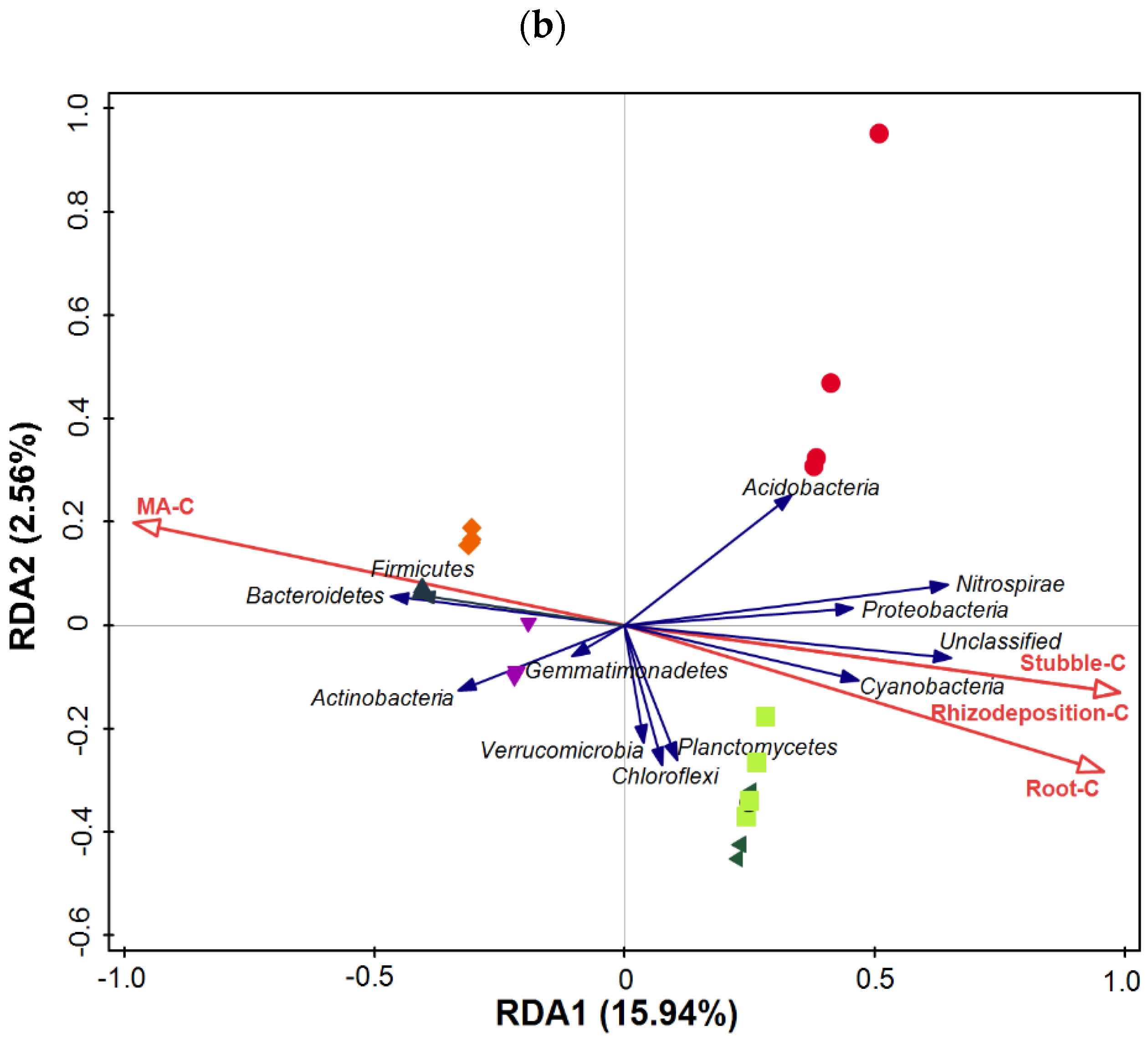
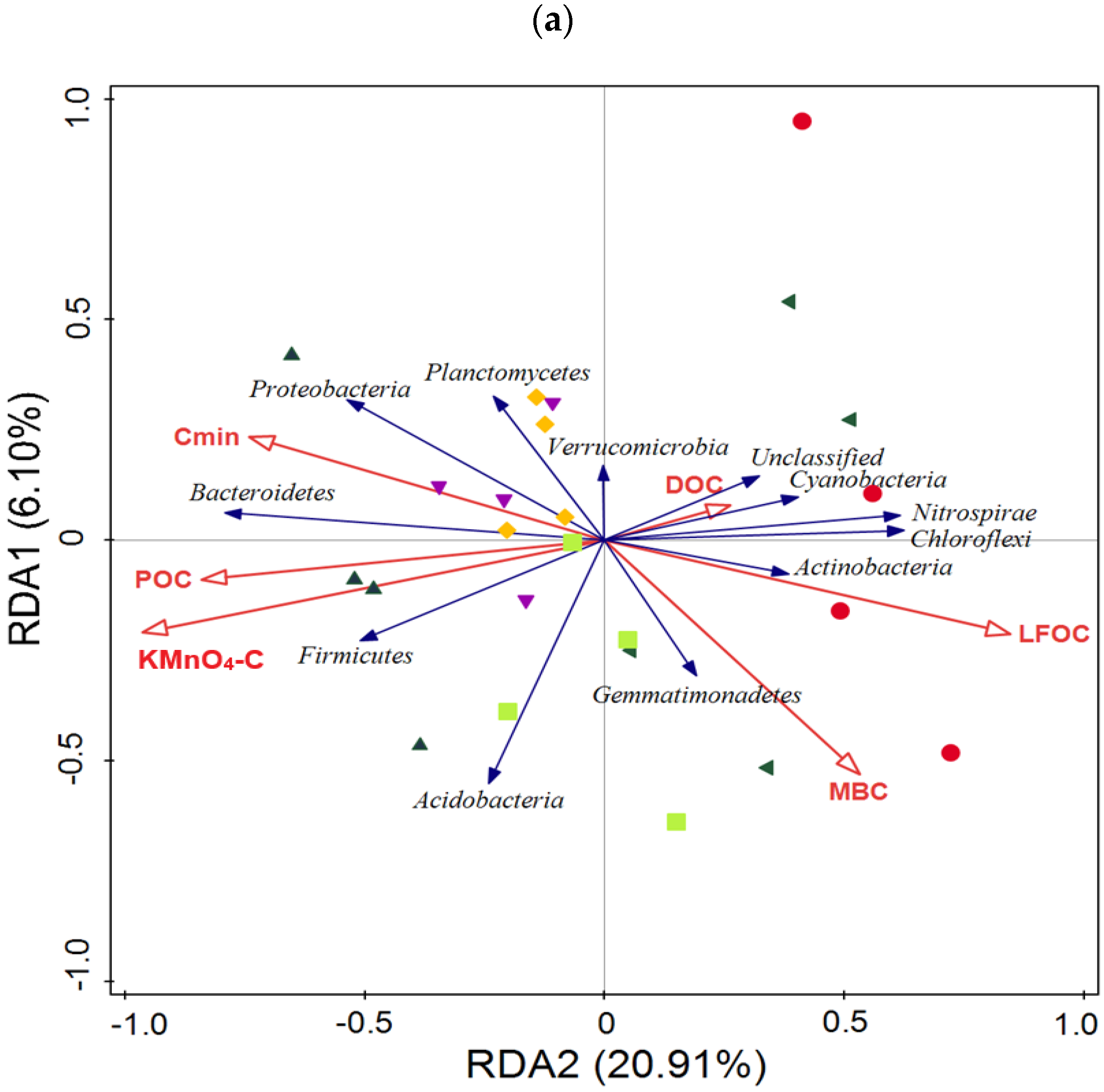
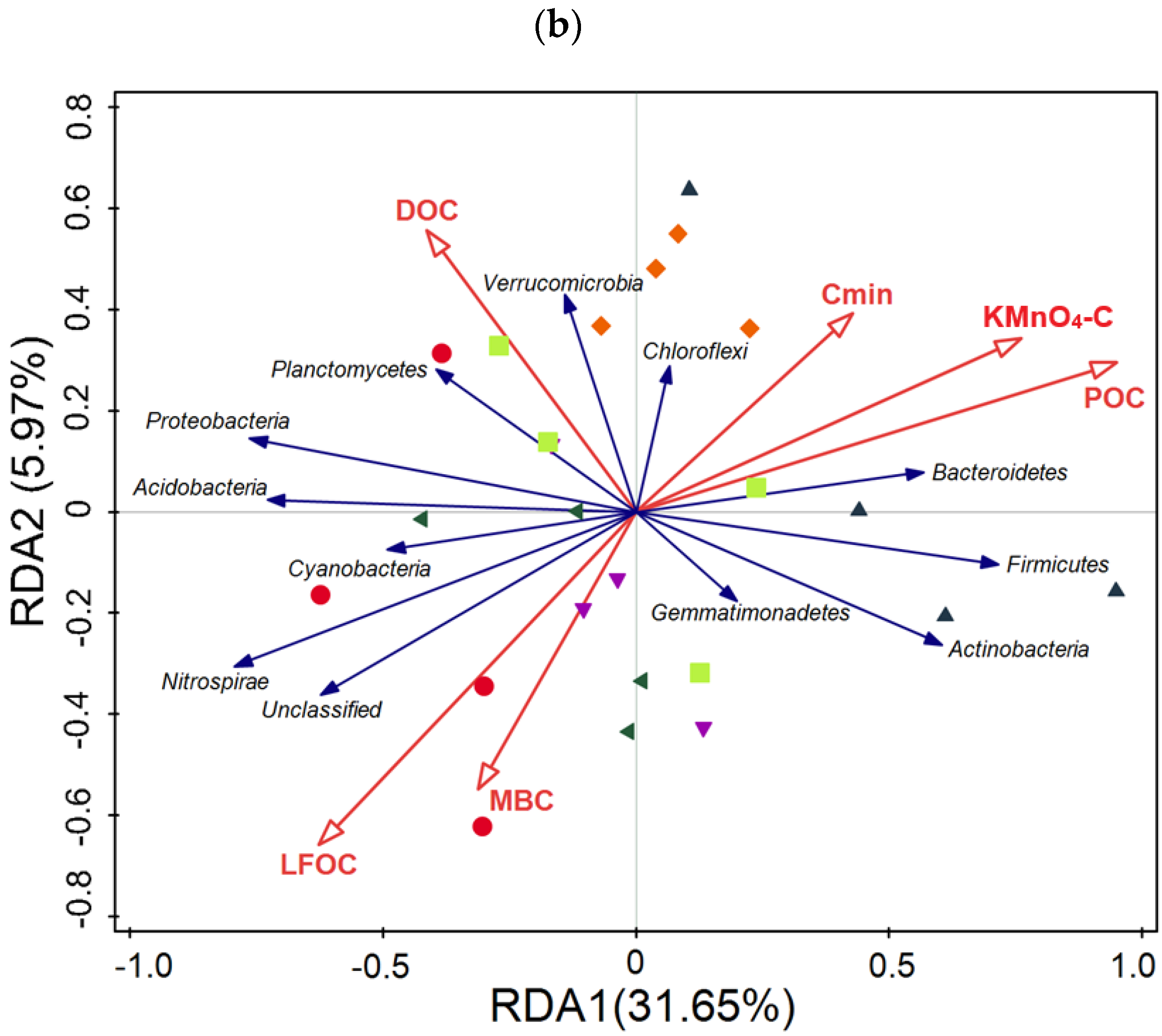
3.5. Taxonomic Composition of Bacterial Communities Compared against Environmental Conditions Affected by Fertilization Regimes
4. Discussion
4.1. Dominant Phyla Reflect Those Commonly Found in Agricultural Soils
4.2. RNA-Based Bacteria Have Lesser Species Diversity and Evenness but Greater Functional Connections Compared with DNA-Based Bacteria
4.3. Significant Effects of Long-Term Fertilization on Bacterial Communities
4.4. Specific Bacterial Taxa in the OM-Fertilized Treatment Compared with Other Treatments
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Pretty, J.; Bharucha, Z.P. Sustainable intensification in agricultural systems. Ann. Bot. 2014, 114, 1571–1596. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Cui, Z.; Fan, M.; Vitousek, P.; Zhao, M.; Ma, W.; Wang, Z.; Zhang, W.; Yan, X.; Yang, J.; et al. Producing more grain with lower environmental costs. Nature 2014, 514, 486–489. [Google Scholar] [CrossRef] [PubMed]
- Luo, G.; Li, L.; Friman, V.-P.; Guo, J.; Guo, S.; Shen, Q.; Ling, N. Organic amendments increase crop yields by improving microbe-mediated soil functioning of agroecosystems: A meta-analysis. Soil Biol. Biochem. 2018, 124, 105–115. [Google Scholar] [CrossRef]
- Van Diepeningen, A.D.; de Vos, O.J.; Korthals, G.W.; van Bruggen, A.H. Effects of organic versus conventional management on chemical and biological parameters in agricultural soils. Appl. Soil Ecol. 2006, 31, 120–135. [Google Scholar] [CrossRef]
- Orr, C.H.; James, A.; Leifert, C.; Cooper, J.M.; Cummings, S.P. Diversity and activity of free-living nitrogen-fixing bacteria and total bacteria in organic and conventionally managed soils. Appl. Environ. Microbiol. 2011, 77, 911–919. [Google Scholar] [CrossRef] [Green Version]
- Chadwick, D.; Wei, J.; Yan’an, T.; Guanghui, Y.; Qirong, S.; Qing, C. Improving manure nutrient management towards sustainable agricultural intensification in China. Agric. Ecosyst. Environ. 2015, 209, 34–46. [Google Scholar] [CrossRef]
- Chen, Q.L.; Ding, J.; Zhu, D.; Hu, H.W.; Delgado Baquerizo, M.; Ma, Y.B.; He, J.Z.; Zhu, Y.G. Rare microbial taxa as the major drivers of ecosystem multifunctionality in long-term fertilized soils. Soil Biol. Biochem. 2020, 141, 107686. [Google Scholar] [CrossRef]
- Sradnick, A.; Murugan, R.; Oltmanns, M.; Raupp, J.; Joergensen, R.G. Changes in functional diversity of the soil microbial community in a heterogeneous sandy soil after long-term fertilization with cattle manure and mineral fertilizer. Appl. Soil Ecol. 2013, 63, 23–28. [Google Scholar] [CrossRef]
- Liu, J.; Shu, A.J.; Song, W.F.; Shi, W.C.; Li, M.C.; Zhang, W.X.; Li, Z.Z.; Liu, G.R.; Yuan, F.S.; Zhang, S.X.; et al. Long-term organic fertilizer substitution increases rice yield by improving soil properties and regulating soil bacteria. Geoderma 2021, 404, 115287. [Google Scholar] [CrossRef]
- Orr, C.H.; Stewart, C.J.; Leifert, C.; Cooper, J.M.; Cummings, S.P. Effect of crop management and sample year on abundance of soil bacterial communities in organic and conventional cropping systems. J. Appl. Microbiol. 2015, 119, 208–214. [Google Scholar] [CrossRef]
- Dai, Z.; Su, W.; Chen, H.; Barberán, A.; Zhao, H.; Yu, M.; Yu, L.; Brookes, P.C.; Schadt, C.W.; Chang, S.X.; et al. Long-term nitrogen fertilization decreases bacterial diversity and favors the growth of Actinobacteria and Proteobacteria in agro-ecosystems across the globe. Glob. Chang. Biol. 2018, 24, 3452–3461. [Google Scholar] [CrossRef] [PubMed]
- Huang, R.; McGrath, S.P.; Hirsch, P.R.; Clark, I.M.; Storkey, J.; Wu, L.; Zhou, J.; Liang, Y. Plant–microbe networks in soil are weakened by century-long use of inorganic fertilizers. Microb. Biotechnol. 2019, 12, 1464–1475. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ling, N.; Sun, Y.; Ma, J.; Guo, J.; Zhu, P.; Peng, C.; Yu, G.; Ran, W.; Guo, S.; Shen, Q. Response of the bacterial diversity and soil enzyme activity in particle-size fractions of Mollisol after different fertilization in a long-term experiment. Biol. Fertil. Soils 2014, 50, 901–911. [Google Scholar] [CrossRef]
- Feng, Y.; Chen, R.; Hu, J.; Fei, Z.; Wang, J.; Chu, H.; Zhang, J.; Dolfing, J.; Lin, X. Bacillus asahii comes to the fore in organic manure fertilized alkaline soils. Soil Biol. Biochem. 2015, 81, 186–194. [Google Scholar] [CrossRef] [Green Version]
- Wang, W.; Wang, H.; Feng, Y.; Wang, L.; Xiao, X.; Xi, Y.; Luo, X.; Sun, R.; Ye, X.; Huang, Y.; et al. Consistent responses of the microbial community structure to organic farming along the middle and lower reaches of the Yangtze River. Sci. Rep. 2016, 6, 35046. [Google Scholar] [CrossRef] [Green Version]
- Bonanomi, G.; De Filippis, F.; Cesarano, G.; La Storia, A.; Ercolini, D.; Scala, F. Organic farming induces changes in soil microbiota that affect agro-ecosystem functions. Soil Biol. Biochem. 2016, 103, 327–336. [Google Scholar] [CrossRef]
- Cookson, W.R.; Abaye, D.A.; Marschner, P.; Murphy, D.V.; Stockdale, E.A.; Goulding, K.W.T. The contribution of soil organic matter fractions to carbon and nitrogen mineralization and microbial community size and structure. Soil Biol. Biochem. 2005, 37, 1726–1737. [Google Scholar] [CrossRef]
- Ramírez, P.B.; Fuentes-Alburquenque, S.; Díez, B.; Vargas, I.; Bonilla, C.A. Soil microbial community responses to labile organic carbon fractions in relation to soil type and land use along a climate gradient. Soil Biol. Biochem. 2020, 141, 107692. [Google Scholar] [CrossRef]
- Li, J.; Cooper, J.M.; Lin, Z.A.; Li, Y.; Yang, X.; Zhao, B. Soil microbial community structure and function are significantly affected by long-term organic and mineral fertilization regimes in the North China Plain. Appl. Soil Ecol. 2015, 96, 75–87. [Google Scholar] [CrossRef]
- Tian, J.; Lou, Y.; Gao, Y.; Fang, H.; Liu, S.; Xu, M.; Blagodatskaya, E.; Kuzyakov, Y. Response of soil organic matter fractions and composition of microbial community to long-term organic and mineral fertilization. Biol. Fertil. Soils 2017, 53, 523–532. [Google Scholar] [CrossRef]
- Gremion, F.; Chatzinotas, A.; Harms, H. Comparative 16S rDNA and 16S rRNA sequence analysis indicates that Actinobacteria might be a dominant part of the metabolically active bacteria in heavy metal-contaminated bulk and rhizosphere soil. Environ. Microbiol. 2003, 5, 896–907. [Google Scholar] [CrossRef] [PubMed]
- Toju, H.; Peay, K.G.; Yamamichi, M.; Narisawa, K.; Hiruma, K.; Naito, K.; Fukuda, S.; Ushio, M.; Nakaoka, S.; Onoda, Y.; et al. Core microbiomes for sustainable agroecosystems. Nat. Plants 2018, 4, 247–257. [Google Scholar] [CrossRef]
- Graham, E.B.; Wieder, W.R.; Leff, J.W.; Weintraub, S.R.; Townsend, A.R.; Cleveland, C.C.; Philippot, L.; Nemergut, D.R. Do we need to understand microbial communities to predict ecosystem function? A comparison of statistical models of nitrogen cycling processes. Soil Biol. Biochem. 2014, 68, 279–282. [Google Scholar] [CrossRef]
- Widder, S.; Allen, R.; Pfeiffer, T.; Curtis, T.; Wiuf, C.; Sloan, W.; Cordero, O.; Brown, S.; Momeni, B.; Shou, W.; et al. Challenges in microbial ecology: Building predictive understanding of community function and dynamics. ISME J. 2016, 10, 2557–2568. [Google Scholar] [CrossRef] [Green Version]
- Cardoso, D.C.; Sandionigi, A.; Cretoiu, M.S.; Casiraghi, M.; Stal, L.; Bolhuis, H. Comparison of the active and resident community of a coastal microbial mat. Sci. Rep. 2017, 7, 2969. [Google Scholar] [CrossRef] [PubMed]
- Che, R.; Deng, Y.; Wang, F.; Wang, W.; Xu, Z.; Wang, Y.; Cui, X. 16S rRNA-based bacterial community structure is a sensitive indicator of soil respiration activity. J. Soil Sediments 2015, 15, 1987–1990. [Google Scholar] [CrossRef]
- Freedman, Z.B.; Romanowicz, K.J.; Upchurch, R.A.; Zak, D.R. Differential responses of total and active soil microbial communities to long-term experimental N deposition. Soil Biol. Biochem. 2015, 90, 275–282. [Google Scholar] [CrossRef] [Green Version]
- Che, R.; Deng, Y.; Wang, W.; Rui, Y.; Zhang, J.; Tahmasbian, I.; Tang, L.; Wang, S.; Wang, Y.; Xu, Z.; et al. Long-term warming rather than grazing significantly changed total and active soil procaryotic community structures. Geoderma 2018, 316, 1–10. [Google Scholar] [CrossRef]
- Kuramae, E.E.; Verbruggen, E.; Hillekens, R.; de Hollander, M.; Röling, W.F.M.; van der Heijden, M.G.A.; Kowalchuk, G.A. Tracking Fungal Community Responses to Maize Plants by DNA- and RNA-based Pyrosequencing. PLoS ONE 2013, 8, e69973. [Google Scholar] [CrossRef] [Green Version]
- Baldrian, P.; Kolarik, M.; Stursova, M.; Kopecky, J.; Valaskova, V.; Vetrovsky, T.; Zifcakova, L.; Snajdr, J.; Ridl, J.; Vlcek, C.; et al. Active and total microbial communities in forest soil are largely different and highly stratified during decomposition. ISME J. 2012, 6, 248–258. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.; Wang, E.; Yang, X.; Zhang, F.; Yin, H. Increased yield potential of wheat-maize cropping system in the North China Plain by climate change adaptation. Clim. Chang. 2012, 113, 825–840. [Google Scholar] [CrossRef]
- Shi, Y.; Li, Y.; Xiang, X.; Sun, R.; Yang, T.; He, D.; Zhang, K.; Ni, Y.; Zhu, Y.-G.; Adams, J.M.; et al. Spatial scale affects the relative role of stochasticity versus determinism in soil bacterial communities in wheat fields across the North China Plain. Microbiome 2018, 6, 27. [Google Scholar] [CrossRef]
- Li, J.; Wen, Y.; Li, X.; Li, Y.; Yang, X.; Lin, Z.; Song, Z.; Cooper, J.M.; Zhao, B. Soil labile organic carbon fractions and soil organic carbon stocks as affected by long-term organic and mineral fertilization regimes in the North China Plain. Soil Tillage Res. 2018, 175, 281–290. [Google Scholar] [CrossRef] [Green Version]
- Mettel, C.; Kim, Y.; Shrestha, P.M.; Liesack, W. Extraction of mRNA from Soil. Appl. Environ. Microbiol. 2010, 76, 5995–6000. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cai, Y.; Zheng, Y.; Bodelier, P.L.E.; Conrad, R.; Jia, Z. Conventional methanotrophs are responsible for atmospheric methane oxidation in paddy soils. Nat. Commun. 2016, 7, 11728. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Amato, K.R.; Yeoman, C.J.; Kent, A.; Righini, N.; Carbonero, F.; Estrada, A.; Rex Gaskins, H.; Stumpf, R.M.; Yildirim, S.; Torralba, M.; et al. Habitat degradation impacts black howler monkey (Alouatta pigra) gastrointestinal microbiomes. ISME J. 2013, 7, 1344–1353. [Google Scholar] [CrossRef]
- Schloss, P.D.; Westcott, S.L.; Ryabin, T.; Hall, J.R.; Hartmann, M.; Hollister, E.B.; Lesniewski, R.A.; Oakley, B.B.; Parks, D.H.; Robinson, C.J.; et al. Introducing mothur: Open-Source, Platform-Independent, Community-Supported Software for Describing and Comparing Microbial Communities. Appl. Environ. Microbiol. 2009, 75, 7537–7541. [Google Scholar] [CrossRef] [Green Version]
- Sun, L.; Xun, W.B.; Huang, T.; Zhang, G.S.; Gao, J.S.; Ran, W.; Li, D.C.; Shen, Q.R.; Zhang, R.F. Alteration of the soil bacterial community during parent material maturation driven by different fertilization treatments. Soil Biol. Biochem. 2016, 96, 207–215. [Google Scholar] [CrossRef]
- Lozupone, C.; Lladser, M.E.; Knights, D.; Stombaugh, J.; Knight, R. UniFrac: An effective distance metric for microbial community comparison. ISME J. 2011, 5, 169–172. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, J.Z.; Deng, Y.; Luo, F.; He, Z.L.; Tu, Q.C.; Zhi, X.Y. Functional molecular ecological networks. mBio 2010, 1, 10. [Google Scholar] [CrossRef] [Green Version]
- Deng, Y.; Jiang, Y.H.; Yang, Y.; He, Z.; Luo, F.; Zhou, J. Molecular ecological network analyses. BMC Bioinform. 2012, 13, 113–132. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ling, N.; Zhu, C.; Xue, C.; Chen, H.; Duan, Y.; Peng, C.; Guo, S.; Shen, Q. Insight into how organic amendments can shape the soil microbiome in long-term field experiments as revealed by network analysis. Soil Biol. Biochem. 2016, 99, 137–149. [Google Scholar] [CrossRef]
- Ogilvie, L.A.; Hirsch, P.R.; Johnston, A.W. Bacterial diversity of the Broadbalk ‘classical’winter wheat experiment in relation to long-term fertilizer inputs. Microb. Ecol. 2008, 56, 525–537. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J.; Ni, T.; Li, Y.; Xiong, W.; Ran, W.; Shen, B.; Shen, Q.; Zhang, R. Responses of bacterial communities in arable soils in a rice-wheat cropping system to different fertilizer regimes and sampling times. PLoS ONE 2014, 9, e85301. [Google Scholar] [CrossRef] [PubMed]
- Janssen, P.H. Identifying the dominant soil bacterial taxa in libraries of 16S rRNA and 16S rRNA genes. Appl. Environ. Microbiol. 2006, 72, 1719–1728. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schloss, P.D.; Handelsman, J. Toward a census of bacteria in soil. PLoS Comput. Biol. 2006, 2, 786–793. [Google Scholar] [CrossRef] [Green Version]
- Ding, J.; Jiang, X.; Ma, M.; Zhou, B.; Guan, D.; Zhao, B.; Zhou, J.; Cao, F.; Li, L.; Li, J. Effect of 35 years inorganic fertilizer and manure amendment on structure of bacterial and archaeal communities in black soil of northeast China. Appl. Soil Ecol. 2016, 105, 187–195. [Google Scholar] [CrossRef]
- Chu, H.; Fierer, N.; Lauber, C.L.; Caporaso, J.G.; Knight, R.; Grogan, P. Soil bacterial diversity in the Arctic is not fundamentally different from that found in other biomes. Environ. Microbiol. 2010, 12, 2998–3006. [Google Scholar] [CrossRef]
- Žifčáková, L.; Větrovský, T.; Howe, A.; Baldrian, P. Microbial activity in forest soil reflects the changes in ecosystem properties between summer and winter. Environ. Microbiol. 2016, 18, 288–301. [Google Scholar] [CrossRef]
- Delgado Baquerizo, M.; Oliverio, A.M.; Brewer, T.E.; Benavent-González, A.; Eldridge, D.J.; Bardgett, R.D.; Maestre, F.T.; Singh, B.K.; Fierer, N. A global atlas of the dominant bacteria found in soil. Science 2018, 359, 320–325. [Google Scholar] [CrossRef] [Green Version]
- Wu, T.; Chellemi, D.; Graham, J.; Martin, K.; Rosskopf, E. Comparison of soil bacterial communities under diverse agricultural land management and crop production practices. Microb. Ecol. 2008, 55, 293–310. [Google Scholar] [CrossRef]
- O’Brien, S.L.; Gibbons, S.M.; Owens, S.M.; Hampton-Marcell, J.; Johnston, E.R.; Jastrow, J.D.; Gilbert, J.A.; Meyer, F.; Antonopoulos, D.A. Spatial scale drives patterns in soil bacterial diversity. Environ. Microbiol. 2016, 18, 2039–2051. [Google Scholar] [CrossRef] [Green Version]
- Liang, Y.; Zhang, Y.; Zhou, C.; Li, H.; Kang, X.; Wang, L.; Song, J.; Jiao, N. Cumulative impact of long-term intensive mariculture on total and active bacterial communities in the core sediments of the Ailian Bay, North China. Sci. Total Environ. 2019, 691, 1212–1224. [Google Scholar] [CrossRef]
- Gill, A.S.; Lee, A.; McGuire, K.L. Phylogenetic and functional diversity of total (DNA) and expressed (RNA) bacterial communities in urban green infrastructure bioswale soils. Appl. Environ. Microbiol. 2017, 83, e00287-17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xiong, W.; Li, R.; Ren, Y.; Liu, C.; Zhao, Q.; Wu, H.; Jousset, A.; Shen, Q. Distinct roles for soil fungal and bacterial communities associated with the suppression of vanilla Fusarium wilt disease. Soil Biol. Biochem. 2017, 107, 198–207. [Google Scholar] [CrossRef]
- Langille, M.G.; Zaneveld, J.; Caporaso, J.G.; McDonald, D.; Knights, D.; Reyes, J.A.; Clemente, J.C.; Burkepile, D.E.; Thurber, R.L.V.; Knight, R.; et al. Predictive functional profiling of microbial communities using 16S rRNA marker gene sequences. Nat. Biotechnol. 2013, 31, 814–821. [Google Scholar] [CrossRef]
- Razanamalala, K.; Razafimbelo, T.; Maron, P.A.; Ranjard, L.; Chemidlin, N.; Lelièvre, M.; Dequiedt, S.; Ramaroson, V.H.; Marsden, C.; Becquer, T.; et al. Soil microbial diversity drives the priming effect along climate gradients: A case study in Madagascar. ISME J. 2017, 12, 451–462. [Google Scholar] [CrossRef]
- Yi, J.; Wu, H.Y.; Wu, J.; Deng, C.Y.; Zheng, R.; Chao, Z. Molecular phylogenetic diversity of Bacillus community and its temporal–spatial distribution during the swine manure of composting. Appl. Microbiol. Biotechnol. 2012, 93, 411–421. [Google Scholar] [CrossRef] [PubMed]
- Jones, S.; Lennon, J. Dormancy contributes to the maintenance of microbial diversity. Proc. Natl. Acad. Sci. USA 2010, 107, 5881–5886. [Google Scholar] [CrossRef] [Green Version]
- McCarren, J.; Becker, J.W.; Repeta, D.J.; Shi, Y.; Young, C.R.; Malmstrom, R.R.; Chisholm, S.W.; DeLong, E.F. Microbial community transcriptomes reveal microbes and metabolic pathways associated with dissolved organic matter turnover in the sea. Proc. Natl. Acad. Sci. USA 2010, 107, 16420–16427. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hugenholtz, P.; Goebel, B.M.; Pace, N.R. Impact of culture-independent studies on the emerging phylogenetic view of bacterial diversity. J. Bacteriol. 1998, 180, 4765–4774. [Google Scholar] [CrossRef] [Green Version]
- Jones, R.T.; Robeson, M.S.; Lauber, C.L.; Hamady, M.; Knight, R.; Fierer, N. A comprehensive survey of soil acidobacterial diversity using pyrosequencing and clone library analyses. ISME J. 2009, 3, 442–453. [Google Scholar] [CrossRef]
- Cederlund, H.; Wessén, E.; Enwall, K.; Jones, C.M.; Juhanson, J.; Pell, M.; Philippot, L.; Hallin, S. Soil carbon quality and nitrogen fertilization structure bacterial communities with predictable responses of major bacterial phyla. Appl. Soil Ecol. 2014, 84, 62–68. [Google Scholar] [CrossRef]
- Birrer, S.C.; Dafforn, K.A.; Simpson, S.L.; Kelaher, B.P.; Potts, J.; Scanes, P.; Johnston, E.L. Interactive effects of multiple stressors revealed by sequencing total (DNA) and active (RNA) components of experimental sediment microbial communities. Sci. Total Environ. 2018, 637–638, 1383–1394. [Google Scholar] [CrossRef]
- Eo, J.; Park, K.C. Long-term effects of imbalanced fertilization on the composition and diversity of soil bacterial community. Agric. Ecosyst. Environ. 2016, 231, 176–182. [Google Scholar] [CrossRef]
- Davis, K.E.R.; Sangwan, P.; Janssen, P.H. Acidobacteria, Rubrobacteridae and Chloroflexi are abundant among very slow-growing and mini-colony-forming soil bacteria. Environ. Microbiol. 2011, 13, 798–805. [Google Scholar] [CrossRef] [PubMed]
- Hester, E.R.; Harpenslager, S.F.; van Diggelen, J.M.H.; Lamers, L.L.; Jetten, M.S.M.; Lüke, C.; Lücker, S.; Welte, C.U. Linking nitrogen load to the structure and function of wetland soil and rhizosphere microbial communities. Msystems 2018, 3, e00214-17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xun, W.; Zhao, J.; Xue, C.; Zhang, G.; Ran, W.; Wang, B.; Shen, Q.; Zhang, R. Significant alteration of soil bacterial communities and organic carbon decomposition by different long-term fertilization management conditions of extremely low-productivity arable soil in South China. Environ. Microbiol. 2016, 18, 1907–1917. [Google Scholar] [CrossRef]
- Zarb, J.; Ghorbani, R.; Koocheki, A.; Leifert, C. The importance of microorganisms in organic agriculture. Outlooks Pest Manag. 2005, 16, 52–55. [Google Scholar] [CrossRef] [Green Version]
- Geisseler, D.; Scow, K.M. Long-term effects of mineral fertilizers on soil microorganisms—A review. Soil Biol. Biochem. 2014, 75, 54–63. [Google Scholar] [CrossRef]
- Fierer, N.; Leff, J.W.; Adams, B.J.; Nielsen, U.N.; Bates, S.T.; Lauber, C.L.; Owens, S.; Gilbert, J.A.; Wall, D.H.; Caporaso, J.G. Cross-biome metagenomic analyses of soil microbial communities and their functional attributes. Proc. Natl. Acad. Sci. USA 2012, 109, 21390–21395. [Google Scholar] [CrossRef] [Green Version]
- Zhang, L.; Shi, N.; Fan, J.; Wang, F.; George, T.S.; Feng, G. Arbuscular mycorrhizal fungi stimulate organic phosphate mobilization associated with changing bacterial community structure under field conditions. Environ. Microbiol. 2018, 20, 2639–2651. [Google Scholar] [CrossRef]
- Wang, C.; Liu, D.; Bai, E. Decreasing soil microbial diversity is associated with decreasing microbial biomass under nitrogen addition. Soil Biol. Biochem. 2018, 120, 126–133. [Google Scholar] [CrossRef]
- Rousk, J.; Bååth, E.; Brookes, P.C.; Lauber, C.L.; Lozupone, C.; Caporaso, J.G.; Knight, R.; Fierer, N. Soil bacterial and fungal communities across a pH gradient in an arable soil. ISME J. 2010, 4, 1340–1351. [Google Scholar] [CrossRef] [PubMed]
- Sun, R.B.; Zhang, X.X.; Guo, X.S.; Wang, D.Z.; Chu, H.Y. Bacterial diversity in soils subjected to long-term chemical fertilization can be more stably maintained with the addition of livestock manure than wheat straw. Soil Biol. Biochem. 2015, 88, 9–18. [Google Scholar] [CrossRef]
- Nicol, G.W.; Leininger, S.; Schleper, C.; Prosser, J.I. Influence of soil pH on the diversity, abundance and transcriptional activity of ammonia oxidizing archaea and bacteria. Environ. Microbiol. 2008, 10, 2966–2978. [Google Scholar] [CrossRef] [PubMed]
- Lauber, C.L.; Hamady, M.; Knight, R.; Fierer, N. Pyrosequencing-based assessment of soil pH as a predictor of soil bacterial community structure at the continental scale. Appl. Environ. Microbiol. 2009, 75, 5111–5120. [Google Scholar] [CrossRef] [Green Version]
- Zhou, J.; Guan, D.; Zhou, B.; Zhao, B.; Ma, M.; Qin, J.; Jiang, X.; Chen, S.; Cao, F.; Shen, D.; et al. Influence of 34-years of fertilization on bacterial communities in an intensively cultivated black soil in northeast China. Soil Biol. Biochem. 2015, 90, 42–51. [Google Scholar] [CrossRef]
- Zhou, J.; Jiang, X.; Wei, D.; Zhao, B.S.; Ma, M.C.; Chen, S.F.; Cao, F.M.; Shen, D.L.; Guan, D.W.; Li, J. Consistent effects of nitrogen fertilization on soil bacterial communities in black soils for two crop seasons in China. Sci. Rep. 2017, 7, 3267. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.; Xue, C.; Song, Y.; Wang, L.; Huang, Q.; Shen, Q. Wheat and Rice Growth Stages and Fertilization Regimes Alter Soil Bacterial Community Structure, But Not Diversity. Front. Microbiol. 2016, 7, 1207. [Google Scholar] [CrossRef]
- Liu, X.; Cong, J.; Lu, H.; Xue, Y.; Wang, X.; Li, D.; Zhang, Y. Community structure and elevational distribution pattern of soil Actinobacteria in alpine grasslands. Acta Ecol. Sin. 2017, 37, 213–218. [Google Scholar] [CrossRef]
- Acosta-Martínez, V.; Dowd, S.; Sun, Y.; Allen, V. Tag-encoded pyrosequencing analysis of bacterial diversity in a single soil type as affected by management and land use. Soil Biol. Biochem. 2008, 40, 2762–2770. [Google Scholar] [CrossRef]
- De Menezes, A.B.; Prendergast-Miller, M.T.; Poonpatana, P.; Farrell, M.; Bissett, A.; Macdonald, L.M.; Toscas, P.; Richardson, A.E.; Thrall, P.H. C/N ratio drives soil actinobacterial cellobiohydrolase gene diversity. Appl. Environ. Microbiol. 2015, 81, 3016–3028. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Daims, H.; Lebedeva, E.V.; Pjevac, P.; Han, P.; Herbold, C.; Albertsen, M.; Jehmlich, N.; Palatinszky, M.; Vierheilig, J.; Bulaev, A.; et al. Complete nitrification by Nitrospira bacteria. Nature 2015, 528, 504–509. [Google Scholar] [CrossRef]
- Coolon, J.; Jones, K.; Todd, T.; Blair, J.; Herman, M. Long-Term Nitrogen Amendment Alters the Diversity and Assemblage of Soil Bacterial Communities in Tallgrass Prairie. PLoS ONE 2013, 8, e67884. [Google Scholar] [CrossRef] [PubMed]
- Ramirez, K.S.; Craine, J.M.; Fierer, N. Consistent effects of nitrogen amendments on soil microbial communities and processes across biomes. Glob. Chang. Biol. 2012, 18, 1918–1927. [Google Scholar] [CrossRef]
- Leite, M.F.A.; Yao, P.; Bloem, J.; Berge, H.T.; Kuramae, E.E. Organic nitrogen rearranges both structure and activity of the soil-borne microbial seedbank. Sci. Rep. 2017, 7, 42634. [Google Scholar] [CrossRef]
- Lin, X.; Feng, Y.; Zhang, H.; Chen, R.; Wang, J.; Zhang, J.; Chu, H. Long-term balanced fertilization decreases arbuscular mycorrhizal fungal diversity in an arable soil in North China revealed by 454 pyrosequencing. Environ. Sci. Technol. 2012, 46, 5764–5771. [Google Scholar] [CrossRef]
- Banerjee, S.; Schlaeppi, K.; van der Heijden, M.G.A. Keystone taxa as drivers of microbiome structure and functioning. Nat. Rev. Microbiol. 2018, 16, 567–576. [Google Scholar] [CrossRef] [PubMed]




 SF: mineral fertilizer input; ◆ SM: cattle manure input; ▼ FM: half mineral fertilizer input plus half manure applied; ■ DF: double amount mineral fertilizer input; ▲ DM: double amount cattle manure input).
SF: mineral fertilizer input; ◆ SM: cattle manure input; ▼ FM: half mineral fertilizer input plus half manure applied; ■ DF: double amount mineral fertilizer input; ▲ DM: double amount cattle manure input).
SF: mineral fertilizer input; ◆ SM: cattle manure input; ▼ FM: half mineral fertilizer input plus half manure applied; ■ DF: double amount mineral fertilizer input; ▲ DM: double amount cattle manure input).
SF: mineral fertilizer input; ◆ SM: cattle manure input; ▼ FM: half mineral fertilizer input plus half manure applied; ■ DF: double amount mineral fertilizer input; ▲ DM: double amount cattle manure input).

 SF: mineral fertilizer input; ◆ SM: cattle manure input; ▼ FM: half mineral fertilizer input plus half manure applied; ■ DF: double amount mineral fertilizer input; ▲ DM: double amount cattle manure input).
SF: mineral fertilizer input; ◆ SM: cattle manure input; ▼ FM: half mineral fertilizer input plus half manure applied; ■ DF: double amount mineral fertilizer input; ▲ DM: double amount cattle manure input).
SF: mineral fertilizer input; ◆ SM: cattle manure input; ▼ FM: half mineral fertilizer input plus half manure applied; ■ DF: double amount mineral fertilizer input; ▲ DM: double amount cattle manure input).
SF: mineral fertilizer input; ◆ SM: cattle manure input; ▼ FM: half mineral fertilizer input plus half manure applied; ■ DF: double amount mineral fertilizer input; ▲ DM: double amount cattle manure input).

 SF: mineral fertilizer input; ◆ SM: cattle manure input; ▼ FM: half mineral fertilizer input plus half manure applied; ■ DF: double amount mineral fertilizer input; ▲ DM: double amount cattle manure input).
SF: mineral fertilizer input; ◆ SM: cattle manure input; ▼ FM: half mineral fertilizer input plus half manure applied; ■ DF: double amount mineral fertilizer input; ▲ DM: double amount cattle manure input).
SF: mineral fertilizer input; ◆ SM: cattle manure input; ▼ FM: half mineral fertilizer input plus half manure applied; ■ DF: double amount mineral fertilizer input; ▲ DM: double amount cattle manure input).
SF: mineral fertilizer input; ◆ SM: cattle manure input; ▼ FM: half mineral fertilizer input plus half manure applied; ■ DF: double amount mineral fertilizer input; ▲ DM: double amount cattle manure input).


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Treatments (T) | Total OTUs | ACE | Chao1 | Shannon | Simpson | |
|---|---|---|---|---|---|---|
| DNA | CK-D | 2819 ± 127 bc | 4703 ± 115 c | 4547 ± 50 b | 7.29 ± 0.13 ab | 0.0017 ± 0.0006 ab |
| FM-D | 2998 ± 192 ab | 5663 ± 661 a | 4823 ± 99 ab | 7.37 ± 0.13 a | 0.0016 ± 0.0007 ab | |
| SM-D | 2840 ± 160 abc | 4770 ± 276 bc | 4625 ± 271 ab | 7.30 ± 0.04 ab | 0.0015 ± 0.0000 ab | |
| SF-D | 3019 ± 101 ab | 4891 ± 189 bc | 4707 ± 192 ab | 7.38 ± 0.06 a | 0.0012 ± 0.0002 b | |
| DM-D | 2485 ± 199 c | 4064 ± 299 c | 3874 ± 316 c | 7.08 ± 0.12 b | 0.0021 ± 0.0004 a | |
| DF-D | 3052 ± 172 a | 5394 ± 671 ab | 4864 ± 243 a | 7.38 ± 0.08 a | 0.0013 ± 0.0002 b | |
| T-D | p < 0.05 | p < 0.05 | p < 0.01 | 0.1278 | 0.0819 | |
| RNA | CK-R | 2644 ± 266 a | 4145 ± 399 bc | 4046 ± 417 ab | 7.31 ± 0.11 a | 0.0012 ± 0.0001 a |
| FM-R | 2735 ± 299 a | 4843 ± 399 a | 4235 ± 339 a | 7.30 ± 0.12 a | 0.0013 ± 0.0004 a | |
| SM-R | 2586 ± 226 a | 4323 ± 408 ab | 4005 ± 295 ab | 7.23 ± 0.11 ab | 0.0014 ± 0.0003 a | |
| SF-R | 2659 ± 217 a | 4237 ± 172 bc | 4074 ± 226 ab | 7.28 ± 0.09 ab | 0.0014 ± 0.0002 a | |
| DM-R | 2468 ± 156 a | 3640 ± 397 c | 3546 ± 342 b | 7.16 ± 0.12 ab | 0.0028 ± 0.0005 a | |
| DF-R | 2627 ± 351 a | 4415 ± 415 ab | 4022 ± 298 ab | 7.13 ± 0.07 b | 0.0027 ± 0.0007 a | |
| T-R | 0.8388 | p < 0.05 | 0.3210 | 0.1990 | 0.0755 | |
| N (Nucleic Acid) | DNA average | 2869 ± 211 a | 4914 ± 562 a | 4573 ± 363 a | 7.30 ± 0.12 a | 0.0016 ± 0.0003 a |
| RNA average | 2620 ± 89 b | 4267 ± 391 b | 3988 ± 232 b | 7.24 ± 0.08 a | 0.0018 ± 0.0007 a | |
| N | p < 0.05 | p < 0.05 | p < 0.01 | 0.2743 | 0.4948 |
| Network Metrics | Groups | |
|---|---|---|
| DNA | RNA | |
| Number of nodes | 67 | 57 |
| Total number of edges | 730 | 643 |
| Number of positive correlations | 0.58 | 0.73 |
| Number of negative correlations | 0.42 | 0.27 |
| Average path length | 1.719 | 1.616 |
| Average clustering coefficient | 0.517 | 0.567 |
| Average degree | 21.791 | 22.561 |
| Connect components | 1 | 1 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, J.; Wen, Y.; Yang, X. Understanding the Responses of Soil Bacterial Communities to Long-Term Fertilization Regimes Using DNA and RNA Sequencing. Agronomy 2021, 11, 2425. https://doi.org/10.3390/agronomy11122425
Li J, Wen Y, Yang X. Understanding the Responses of Soil Bacterial Communities to Long-Term Fertilization Regimes Using DNA and RNA Sequencing. Agronomy. 2021; 11(12):2425. https://doi.org/10.3390/agronomy11122425
Chicago/Turabian StyleLi, Juan, Yanchen Wen, and Xiangdong Yang. 2021. "Understanding the Responses of Soil Bacterial Communities to Long-Term Fertilization Regimes Using DNA and RNA Sequencing" Agronomy 11, no. 12: 2425. https://doi.org/10.3390/agronomy11122425