
Bioinformatic-Based Approaches for Disease-Resistance Gene Discovery in Plants


Abstract
:1. Introduction
2. Traditional Map-Based Cloning
3. Bioinformatic-Based Approaches and Pipelines
3.1. NLR Annotation Tools
3.1.1. NLR-Parser
3.1.2. NLR-Annotator
3.1.3. DRAGO2
3.1.4. NLGenomeSweeper
3.1.5. RRGPredictor
3.1.6. NLRtracker
3.2. NLR Discovering Pipelines
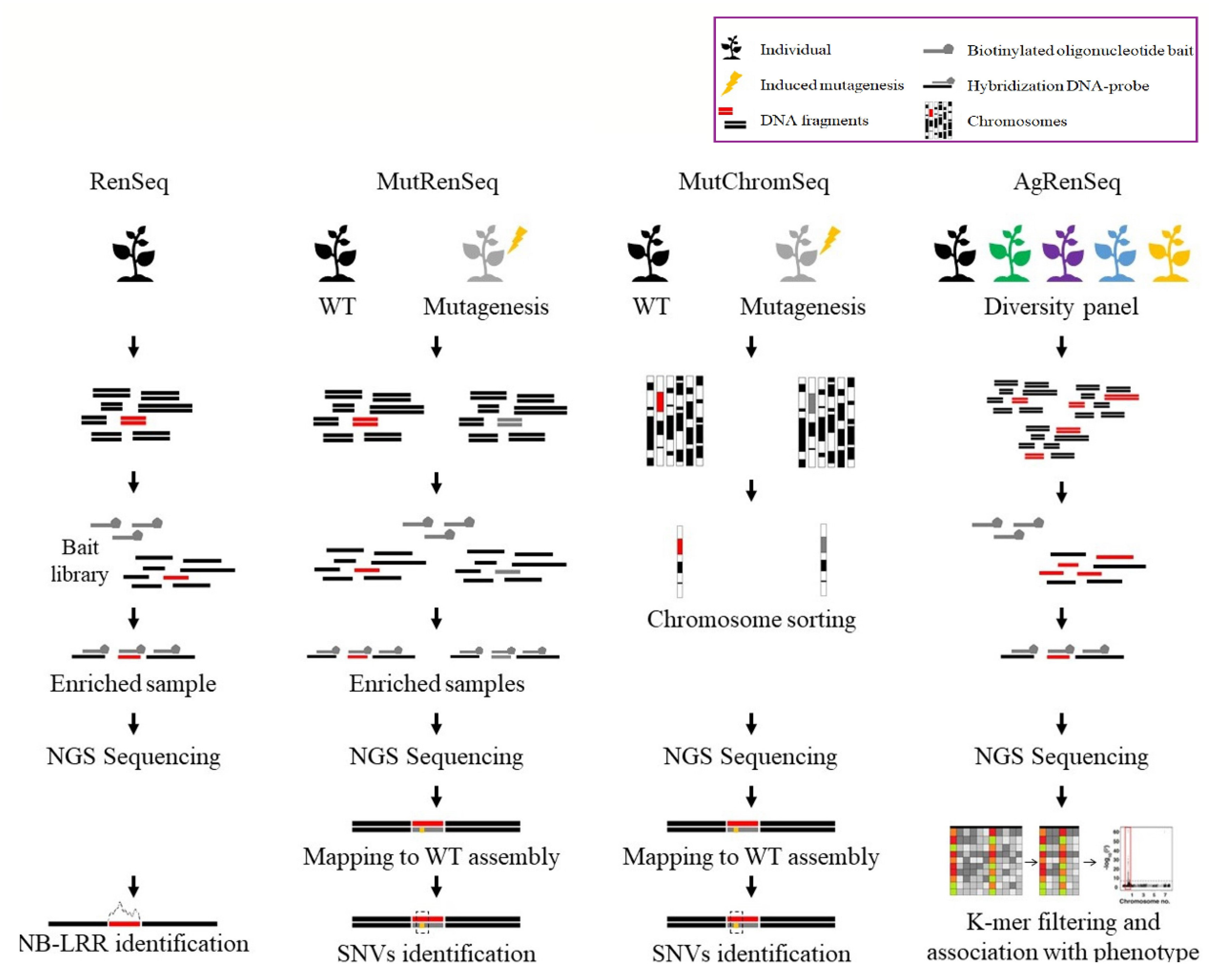
3.2.1. RenSeq
3.2.2. MutRenSeq
3.2.3. MutChromSeq
3.2.4. AgRenSeq
4. Remarks and Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Mcdonald, B.A.; Stukenbrock, E.H. Rapid emergence of pathogens in agro-ecosystems: Global threats to agricultural sustainability and food security. Philos. Trans. R. Soc. B Biol. Sci. 2016, 371, 20160026. [Google Scholar] [CrossRef] [Green Version]
- Viruel, J.; Kantar, M.B.; Gargiulo, R.; Hesketh-Prichard, P.; Leong, N.; Cockel, C.; Forest, F.; Gravendeel, B.; Pérez-Barrales, R.; Leitch, I.J.; et al. Crop wild phylorelatives (CWPs): Phylogenetic distance, cytogenetic compatibility and breeding system data enable estimation of crop wild relative gene pool classification. Bot. J. Linn. Soc. 2021, 195, 1–33. [Google Scholar] [CrossRef]
- Jones, J.; Dangl, J. The plant immune system. Nature 2006, 444, 323–329. [Google Scholar] [CrossRef] [Green Version]
- Kumar, J.; Ramlal, A.; Kumar, K.; Rani, A.; Mishra, V. Signaling Pathways and Downstream Effectors of Host Innate Immunity in Plants. Int. J. Mol. Sci. 2021, 22, 9022. [Google Scholar] [CrossRef]
- Cesari, S.; Bernoux, M.; Moncuquet, P.; Kroj, T.; Dodds, P.N. A novel conserved mechanism for plant NLR protein pairs: The “integrated decoy” hypothesis. Front. Plant Sci. 2014, 5, 606. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kourelis, J.; Sakai, T.; Adachi, H.; Kamoun, S. RefPlantNLR: A comprehensive collection of experimentally validated plant NLRs. BioRxiv 2020. [Google Scholar] [CrossRef]
- Smith, S.M.; Pryor, A.J.; Hulbert, S.H. Allelic and Haplotypic Diversity at the Rp1 Rust Resistance Locus of Maize. Genetics 2004, 167, 1939–1947. [Google Scholar] [CrossRef] [Green Version]
- Calicioglu, O.; Flammini, A.; Bracco, S.; Bellù, L.; Sims, R. The Future Challenges of Food and Agriculture: An Integrated Analysis of Trends and Solutions. Sustainability 2019, 11, 222. [Google Scholar] [CrossRef] [Green Version]
- Myers, S.S.; Smith, M.R.; Guth, S.; Golden, C.D.; Vaitla, B.; Mueller, N.D.; Dangour, A.D.; Huybers, P. Climate Change and Global Food Systems: Potential Impacts on Food Security and Undernutrition. Annu. Rev. Public Health 2017, 382, 59–77. [Google Scholar] [CrossRef]
- Kamatham, S.; Munagapati, S.; Manikanta, K.N.; Vulchi, R.; Chadipiralla, K.; Indla, S.H.; Allam, U.S. Recent advances in engineering crop plants for resistance to insect pests. Egypt. J. Biol. Pest Control 2021, 31, 120. [Google Scholar] [CrossRef]
- van Wersch, S.; Tian, L.; Hoy, R.; Li, X. Plant NLRs: The Whistleblowers of Plant Immunity. Plant Commun. 2020, 1, 100016. [Google Scholar] [CrossRef]
- Gutierrez-Gonzalez, J.J.; Garvin, D.F. De Novo Transcriptome Assembly in Polyploid Species; Eds Gasparis, Sebastian. Oat Methods Protoc. 2017, 1536, 209–221. [Google Scholar] [CrossRef]
- The International Wheat Genome Sequencing Consortium (IWGSC). Shifting the limits in wheat research and breeding using a fully annotated reference genome. Science. 2018, 361, eaar7191. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gutierrez-Gonzalez, J.J.; Mascher, M.; Poland, J.; Muehlbauer, G.J. Dense genotyping-by-sequencing linkage maps of two Synthetic W7984×Opata reference populations provide insights into wheat structural diversity. Sci. Rep. 2019, 9, 1793. [Google Scholar] [CrossRef] [PubMed]
- Walkowiak, S.; Gao, L.; Monat, C.; Haberer, G.; Kassa, M.T.; Brinton, J.; Ramirez-Gonzalez, R.H.; Kolodziej, M.C.; Delorean, E.; Thambugala, D.; et al. Multiple wheat genomes reveal global variation in modern breeding. Nature 2020, 588, 277–283. [Google Scholar] [CrossRef]
- Gutierrez-Gonzalez, J.J.; Garvin, D.F. Subgenome-specific assembly of vitamin E biosynthesis genes and expression patterns during seed development provide insight into the evolution of oat genome. Plant Biotechnol. J. 2016, 14, 2147–2157. [Google Scholar] [CrossRef]
- Brueggeman, R.; Druka, A.; Nirmala, J.; Cavileer, T.; Drader, T.; Rostoks, N.; Mirlohi, A.; Bennypaul, H.; Gill, U.; Kudrna, D.; et al. The stem rust resistance gene Rpg5 encodes a protein with nucleotide-binding-site, leucine-rich, and protein kinase domains. Proc. Natl. Acad. Sci. USA 2008, 105, 14970–14975. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, X.; Richards, J.; Gross, T.; Druka, A.; Kleinhofs, A.; Steffenson, B.; Acevedo, M.; Brueggeman, R. The rpg4-mediated resistance to wheat stem rust (Puccinia graminis) in barley (Hordeum vulgare) requires Rpg5, a second NBS-LRR gene, and an actin depolymerization factor. Mol. Plant Microbe Interact. 2013, 26, 407–418. [Google Scholar] [CrossRef] [Green Version]
- Periyannan, S.; Moore, J.; Ayliffe, M.; Bansal, U.; Wang, X.; Huang, L.; Deal, K.; Luo, M.; Kong, X.; Bariana, H.; et al. The Gene Sr33, an Ortholog of Barley Mla Genes, Encodes Resistance to Wheat Stem Rust Race Ug99. Science 2013, 341, 786–788. [Google Scholar] [CrossRef] [PubMed]
- Thind, A.K.; Wicker, T.; Krattinger, S.G. Rapid Identification of Rust Resistance Genes Through Cultivar-Specific De Novo Chromosome Assemblies. Methods Mol. Biol. 2017, 1659, 245–255. [Google Scholar] [CrossRef]
- Thind, A.; Wicker, T.; Šimková, H.; Fossati, D.; Moullet, O.; Brabant, C.; Vrána, J.; Doležel, J.; Krattinger, S.G. Rapid cloning of genes in hexaploid wheat using cultivar-specific long-range chromosome assembly. Nat. Biotechnol. 2017, 35, 793–796. [Google Scholar] [CrossRef] [PubMed]
- Huerta-Espino, J.; Singh, R.P.; Germán, S.; McCallum, B.D.; Park, R.F.; Chen, W.Q.; Bhardwaj, S.C.; Goyeau, H. Global status of wheat leaf rust caused by Puccinia triticina. Euphytica 2011, 179, 143–160. [Google Scholar] [CrossRef]
- Dyck, P.L.; Kerber, E.R. Inheritance in hexaploid wheat of adult-plant leaf rust resistance derived from Aegilops squarrosa. Can. J. Genet. Cytol. 1970, 12, 175–180. [Google Scholar] [CrossRef]
- Hiebert, C.W.; Thomas, J.B.; Somers, D.J.; McCallum, B.D.; Fox, S.L. Microsatellite mapping of adult-plant leaf rust resistance gene Lr22a in wheat. Theor. Appl. Genet. 2007, 115, 877–884. [Google Scholar] [CrossRef] [PubMed]
- Gutierrez-Gonzalez, J.J.; Garvin, D.F. Reference Genome-Directed Resolution of Homologous and Homeologous Relationships within and between Different Oat Linkage Maps. Plant Genome 2011, 4, 178–190. [Google Scholar] [CrossRef] [Green Version]
- Steuernagel, B.; Jupe, F.; Witek, K.; Jones, J.D.; Wulff, B.B. NLR-parser: Rapid annotation of plant NLR complements. Bioinformatics 2015, 31, 1665–1667. [Google Scholar] [CrossRef] [Green Version]
- Steuernagel, B.; Witek, K.; Krattinger, S.G.; Ramirez-Gonzalez, R.H.; Schoonbeek, H.-J.; Yu, G.; Baggs, E.; Witek, A.I.; Yadav, I.; Krasileva, K.V.; et al. The NLR-Annotator Tool Enables Annotation of the Intracellular Immune Receptor Repertoire. Plant Physiol. 2020, 183, 468–482. [Google Scholar] [CrossRef] [Green Version]
- Osuna-Cruz, C.M.; Paytuvi-Gallart, A.; Di Donato, A.; Sundesha, V.; Andolfo, G.; Cigliano, R.A.; Sanseverino, W.; Ercolano, M.R. PRGdb 3.0: A comprehensive platform for prediction and analysis of plant disease resistance genes. Nucleic Acids Res. 2018, 46, D1197–D1201. [Google Scholar] [CrossRef]
- Toda, N.; Rustenholz, C.; Baud, A.; Le Paslier, M.-C.; Amselem, J.; Merdinoglu, D.; Faivre-Rampant, P. NLGenomeSweeper: A Tool for Genome-Wide NBS-LRR Resistance Gene Identification. Genes 2020, 11, 333. [Google Scholar] [CrossRef] [Green Version]
- Silva, R.J.S.; Micheli, F. RRGPredictor, a set-theory-based tool for predicting pathogen-associated molecular pattern receptors (PRRs) and resistance (R) proteins from plants. Genomics 2020, 112, 2666–2676. [Google Scholar] [CrossRef]
- Bailey, T.L.; Johnson, J.; Grant, C.E.; Noble, W.S. The MEME Suite. Nucleic Acids Res. 2015, 43, W39–W49. [Google Scholar] [CrossRef] [Green Version]
- Wang, L.; Zhao, L.; Zhang, X.; Zhang, Q.; Jia, Y.; Wang, G.; Li, S.; Tian, D.; Li, W.H.; Yang, S. Large-scale identification and functional analysis of NLR genes in blast resistance in the Tetep rice genome sequence. Proc. Natl. Acad. Sci. USA 2019, 116, 18479–18487. [Google Scholar] [CrossRef] [Green Version]
- Finn, R.D.; Mistry, J.; Tate, J.; Coggill, P.; Heger, A.; Pollington, J.E.; Gavin, O.L.; Gunasekaran, P.; Ceric, G.; Forslund, K.; et al. The Pfam protein families database. Nucleic Acids Res. 2010, 38, D211–D222. [Google Scholar] [CrossRef]
- Barragan, A.C.; Weigel, D. Plant NLR diversity: The known unknowns of pan-NLRomes. Plant Cell 2021, 33, 814–831. [Google Scholar] [CrossRef]
- Wu, C.-H.; Abd-El-Haliem, A.; Bozkurt, T.O.; Belhaj, K.; Terauchi, R.; Vossen, J.H.; Kamoun, S. NLR network mediates immunity to diverse plant pathogens. Proc. Natl. Acad. Sci. USA 2017, 114, 8113–8118. [Google Scholar] [CrossRef] [Green Version]
- Camacho, C.; Coulouris, G.; Avagyan, V.; Ma, N.; Papadopoulos, J.; Bealer, K.; Madden, T.L. BLAST+: Architecture and applications. BMC Bioinform. 2009, 10, 421. [Google Scholar] [CrossRef] [Green Version]
- Neupane, S.; Andersen, E.J.; Neupane, A.; Nepal, M.P. Genome-Wide Identification of NBS-Encoding Resistance Genes in Sunflower (Helianthus annuus L.). Genes 2018, 9, 384. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Finn, R.D.; Attwood, T.K.; Babbitt, P.C.; Bateman, A.; Bork, P.; Bridge, A.J.; Chang, H.-Y.; Dosztányi, Z.; El-Gebali, S.; Fraser, M.; et al. InterPro in 2017—Beyond protein family and domain annotations. Nucleic Acids Res. 2017, 45, D190–D199. [Google Scholar] [CrossRef] [PubMed]
- Jupe, F.; Pritchard, L.; Etherington, G.J.; MacKenzie, K.; Cock, P.J.A.; Wright, F.; Sharma, S.K.; Bolser, D.; Bryan, G.J.; Jones, J.D.G.; et al. Identification and localisation of the NB-LRR gene family within the potato genome. BMC Genom. 2012, 13, 75. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jupe, F.; Witek, K.; Verweij, W.; Śliwka, J.; Pritchard, L.; Etherington, G.J.; Maclean, D.; Cock, P.J.; Leggett, R.M.; Bryan, G.J.; et al. Resistance gene enrichment sequencing (RenSeq) enables reannotation of the NB-LRR gene family from sequenced plant genomes and rapid mapping of resistance loci in segregating populations. Plant J. 2013, 76, 530–544. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bailey, T.L.; Gribskov, M. Methods and statistics for combining motif match scores. J. Comput. Biol. 1998, 5, 211–221. [Google Scholar] [CrossRef] [PubMed]
- Tomato Genome Consortium (TGC). The tomato genome sequence provides insights into fleshy fruit evolution. Nature 2012, 485, 635–641. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Andolfo, G.; Jupe, F.; Witek, K.; Etherington, G.J.; Ercolano, M.R.; Jones, J.D. Defining the full tomato NB-LRR resistance gene repertoire using genomic and cDNA RenSeq. BMC Plant Biol. 2014, 14, 120. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barbey, C.R.; Lee, S.; Verma, S.; Bird, K.A.; Yocca, A.E.; Edger, P.P.; Knapp, S.J.; Whitaker, V.M.; Folta, K.M. Disease Resistance Genetics and Genomics in Octoploid Strawberry. G3 Genes Genomes Genet. 2019, 9, 3315–3332. [Google Scholar] [CrossRef] [Green Version]
- Brylińska, M.; Tomczyńska, I.; Jakuczun, H.; Wasilewicz-Flis, I.; Witek, K.; Jones, J.D.G.; Śliwka, J. Fine mapping of the Rpi-rzc1 gene conferring broad-spectrum resistance to potato late blight. Eur. J. Plant Pathol. 2015, 143, 193–198. [Google Scholar] [CrossRef] [Green Version]
- Narang, D.; Kaur, S.; Steuernagel, B.; Ghosh, S.; Bansal, U.; Li, J.; Zhang, P.; Bhardwaj, S.; Uauy, C.; Wulff, B.B.H.; et al. Discovery and characterisation of a new leaf rust resistance gene introgressed in wheat from wild wheat Aegilops peregrina. Sci. Rep. 2020, 10, 7573. [Google Scholar] [CrossRef]
- Witek, K.; Jupe, F.; Witek, A.I.; Baker, D.; Clark, M.D.; Jones, J.D.G. Accelerated cloning of a potato late blight–resistance gene using RenSeq and SMRT sequencing. Nat. Biotechnol. 2016, 34, 656–660. [Google Scholar] [CrossRef] [Green Version]
- Van de Weyer, A.L.; Monteiro, F.; Furzer, O.J.; Nishimura, M.T.; Cevik, V.; Witek, K.; Jones, J.D.G.; Dangl, J.L.; Weigel, D.; Bemm, F. A Species-Wide Inventory of NLR Genes and Alleles in Arabidopsis thaliana. Cell 2019, 178, 1260–1272. [Google Scholar] [CrossRef] [Green Version]
- Strachan, S.M.; Armstrong, M.R.; Kaur, A.; Wright, K.M.; Lim, T.Y.; Baker, K.; Jones, J.; Bryan, G.; Blok, V.; Hein, I. Mapping the H2 resistance effective against Globodera pallida pathotype Pa1 in tetraploid potato. Theor. Appl. Genet. 2019, 132, 1283–1294. [Google Scholar] [CrossRef] [Green Version]
- Chen, X.; Lewandowska, D.; Armstrong, M.R.; Baker, K.; Lim, T.-Y.; Bayer, M.; Harrower, B.; McLean, K.; Jupe, F.; Witek, K.; et al. Identification and rapid mapping of a gene conferring broad-spectrum late blight resistance in the diploid potato species Solanum verrucosum through DNA capture technologies. Theor. Appl. Genet. 2018, 131, 1287–1297. [Google Scholar] [CrossRef] [Green Version]
- Steuernagel, B.; Peiyannan, S.K.; Hernández-Pinzón, I.; Witek, K.; Rouse, M.N.; Yu, G.; Hatta, A.; Ayliffe, M.; Bariana, H.; Jones, J.D.G.; et al. Rapid cloning of disease-resistance genes in plants using mutagenesis and sequence capture. Nat. Biotechnol. 2016, 34, 652–655. [Google Scholar] [CrossRef] [PubMed]
- Steuernagel, B.; Witek, K.; Jones, J.D.G.; Wulff, B.B.H. MutRenSeq: A method for rapid cloning of plant disease resistance genes. Methods Mol. Biol. 2017, 1659, 215–229. [Google Scholar] [PubMed]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R. The Sequence Alignment/Map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef] [Green Version]
- Marchal, C.; Zhang, J.; Zhang, P.; Fenwick, P.; Steuernagel, B.; Adamski, N.M.; Boyd, L.; Mclntosh, R.; Wulff, B.B.H.; Berry, S.; et al. BED-domain-containing immune receptors confer diverse resistance spectra to yellow rust. Nat. Plants 2018, 4, 662–668. [Google Scholar] [CrossRef]
- Sánchez-Martín, J.; Steuernagel, B.; Ghosh, S.; Herren, G.; Hurni, S.; Adamski, N.; Vrána, J.; Kubaláková, M.; Krattinger, S.G.; Wicker, T.; et al. Rapid gene isolation in barley and wheat by mutant chromosome sequencing. Genome Biol. 2016, 17, 221. [Google Scholar] [CrossRef] [Green Version]
- Steuernagel, B.; Vrána, J.; Karafiátová, M.; Wulff, B.B.H.; Doležel, J. Rapid Gene Isolation Using MutChromSeq. Methods Mol. Biol. 2017, 1659, 231–243. [Google Scholar] [CrossRef]
- Robinson, J.; Thorvaldsdóttir, H.; Winckler, W.; Guttman, M.; Lander, E.S.; Getz, G.; Mesirov, J.P. Integrative genomics viewer. Nat. Biotechnol. 2011, 29, 24–26. [Google Scholar] [CrossRef] [Green Version]
- Park, R.F.; Golegaonkar, P.G.; Derevnina, L.; Sandhu, K.S.; Karaoglu, H.; Elmansour, H.M.; Dracatos, P.M.; Singh, D. Leaf rust of cultivated barley: Pathology and control. Annu. Rev. Phytopathol. 2015, 53, 565–589. [Google Scholar] [CrossRef] [PubMed]
- Dracatos, P.M.; Barto¡, J.; Elmansour, H.; Singh, D.; Karafiátová, M.; Zhang, P.; Steuernagel, B.; Svačina, R.; Cobbin, J.C.A.; Clark, B.; et al. The Coiled-Coil NLR Rph1, Confers Leaf Rust Resistance in Barley Cultivar Sudan. Plant Physiol. 2019, 179, 1362–1372. [Google Scholar] [CrossRef] [Green Version]
- Sánchez-Martín, J.; Widrig, V.; Herren, G.; Wicker, T.; Zbinden, H.; Gronnier, J.; Spörri, L.; Praz, C.R.; Heuberger, M.; Kolodziej, M.C.; et al. Wheat Pm4 resistance to powdery mildew is controlled by alternative splice variants encoding chimeric proteins. Nat. Plants 2021, 7, 327–341. [Google Scholar] [CrossRef]
- Kolodziej, M.C.; Singla, J.; Sánchez-Martín, J.; Zbinden, H.; Šimková, H.; Karafiátová, M.; Doležel, J.; Gronnier, J.; Poretti, M.; Glauser, G.; et al. A membrane-bound ankyrin repeat protein confers race-specific leaf rust disease resistance in wheat. Nat. Commun. 2021, 12, 956. [Google Scholar] [CrossRef] [PubMed]
- Vo, K.T.X.; Kim, C.Y.; Chandran, A.K.N.; Jung, K.-H.; An, G.; Jeon, J.-S. Molecular insights into the function of ankyrin proteins in plants. J. Plant Biol. 2015, 58, 271–284. [Google Scholar] [CrossRef]
- Schneider, L.M.; Adamski, N.M.; Christensen, C.E.; Stuart, D.B.; Vautrin, S.; Hansson, M.; Uauy, C.; von Wettstein-Knowles, P. The Cer-cqu gene cluster determines three key players in a beta-diketone synthase polyketide pathway synthesizing aliphatics in epicuticular waxes. J. Exp. Bot. 2016, 67, 2715–2730. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arora, S.; Steuernagel, B.; Gaurav, K.; Chandramohan, S.; Long, Y.; Mathy, O.; Johnson, R.; Enk, J.; Periyannan, S.; Singh, N.; et al. Resistance gene cloning from a wild crop relative by sequence capture and association genetics. Nat. Biotechnol. 2019, 37, 139–143. [Google Scholar] [CrossRef] [PubMed]
- Lees, J.; Vehkala, M.; Välimäki, N.; Harris, S.R.; Chewapreecha, C.; Croucher, N.J.; Marttinen, P.; Davies, M.R.; Steer, A.C.; Tong, S.Y.C.; et al. Sequence element enrichment analysis to determine the genetic basis of bacterial phenotypes. Nat. Commun. 2016, 7, 12797. [Google Scholar] [CrossRef]
- Gaurav, K.; Arora, S.; Silva, P.; Sánchez-Martín, J.; Horsnell, R.; Gao, L.; Brar, G.S.; Widrig, V.; Raupp, J.; Singh, N.; et al. Evolution of the bread wheat D-Subgenome and enriching it with diversity from Aegilops tauschii. Biorxiv 2021. [Google Scholar] [CrossRef]


{kind=link}
| Tool | Dependencies | Advantages | Disadvantages | Input | Reference |
|---|---|---|---|---|---|
| NLR-parser | motif alignment and MAST | Discrimination of pseudogenes | Predefined gene models needed | Amino acids | [26] |
| NLR-annotator | meme-suite, NLR-parser | Independent of gene expression, highest domain annotation accuracy, high sensitivity, high specificity | Partial or pseudogenized genes represented, duplication NLRs with multiple NB-ARC domains | Transcript/genomic | [27] |
| DRAGO2 | HMMER, COILS, TMHMM | High sensitivity, web-based interface | Medium domain annotation accuracy | Transcript/amino acids | [28] |
| NLGenome-Sweeper | BLAST+, MUSCLE, SAMtools, bedtools, HMMER, InterProScan, TransDecoder | High specificity, previous gene predictions not required, good performance for RNL genes | Duplication NLRs with multiple NB-ARC domains, low domain annotation accuracy, very high computational cost | Transcript/genomic | [29] |
| RRGPredictor | InterProScan | High specificity, alignment or sequence homology not needed | High computational cost | Transcript/amino acids | [30] |
| NLRtracker | InterProScan | Output of extracted NB-ARC domain, classification NLRs into subgroups, high specificity | Not enough information available | Transcript/amino acids | [6] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fernandez-Gutierrez, A.; Gutierrez-Gonzalez, J.J. Bioinformatic-Based Approaches for Disease-Resistance Gene Discovery in Plants. Agronomy 2021, 11, 2259. https://doi.org/10.3390/agronomy11112259
Fernandez-Gutierrez A, Gutierrez-Gonzalez JJ. Bioinformatic-Based Approaches for Disease-Resistance Gene Discovery in Plants. Agronomy. 2021; 11(11):2259. https://doi.org/10.3390/agronomy11112259
Chicago/Turabian StyleFernandez-Gutierrez, Andrea, and Juan J. Gutierrez-Gonzalez. 2021. "Bioinformatic-Based Approaches for Disease-Resistance Gene Discovery in Plants" Agronomy 11, no. 11: 2259. https://doi.org/10.3390/agronomy11112259