Genome-Wide Diversity Analysis of Valeriana officinalis L. Using DArT-seq Derived SNP Markers

 , , ,
, , ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Plant Materials
2.2. DNA Extraction and Genetic Analysis
2.3. Data Analysis
3. Results
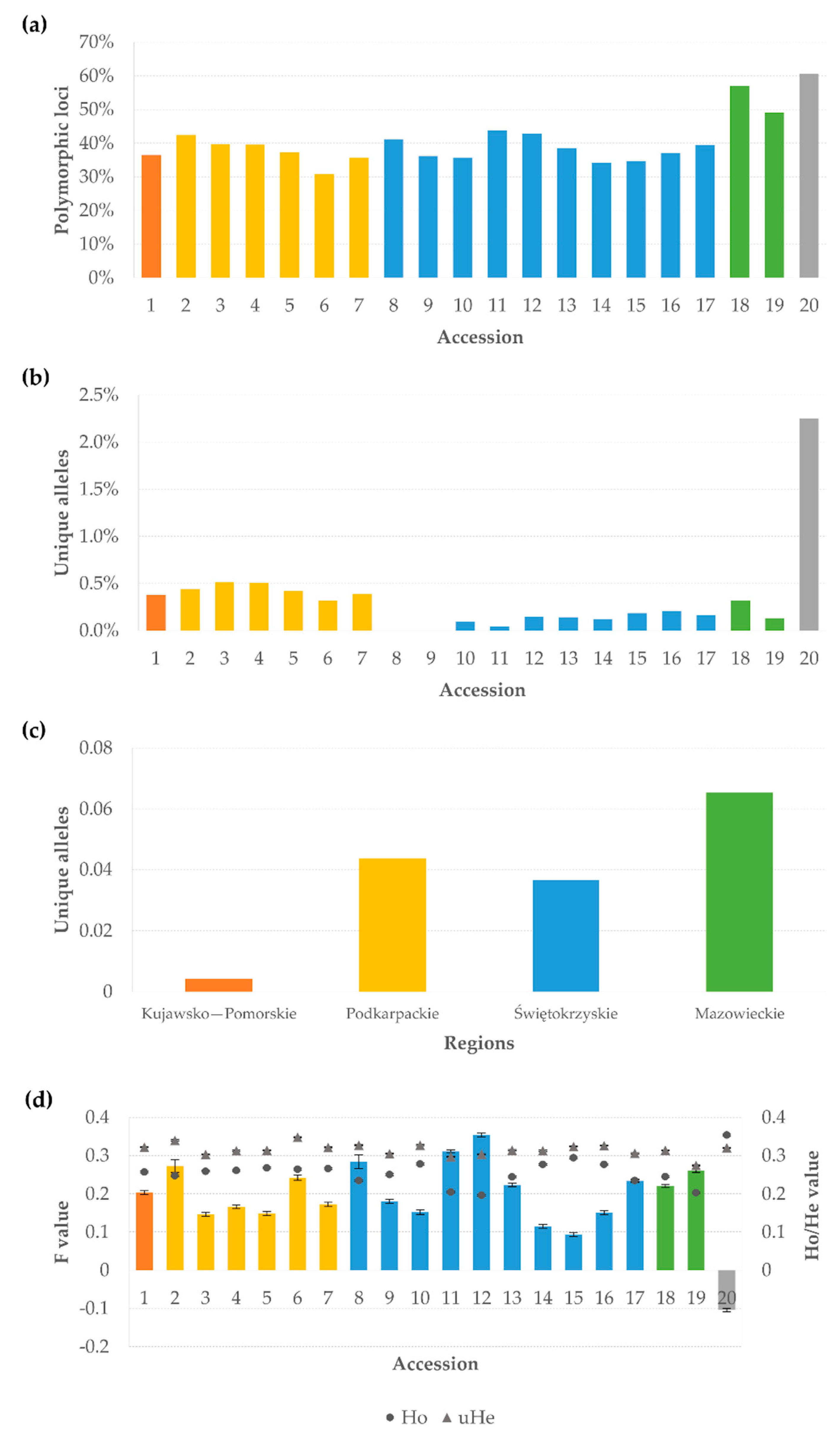
3.1. Marker Quality Analysis
3.2. Genetic Diversity
3.3. Mantel Test
3.4. AMOVA
3.5. Population Structure
4. Discussion
4.1. Utility of DArT-seq Markers
4.2. Diversity
4.3. Gene Pool
4.4. Vulnerabilities
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Patočka, J.; Jakl, J. Biomedically relevant chemical constituents of Valeriana officinalis. J. Appl. Biomed. 2010, 8, 11–18. [Google Scholar] [CrossRef] [Green Version]
- European Pharmacopoeia. Baldrianwurzel Valerianae Radix s.l., 9th ed.; Verlag GmbH: Stuttgart, Germany, 2017. [Google Scholar]
- Wichtl, M. Herbal Drugs and Phytopharmaceuticals: A Handbook for Practice on a Scientific Basis; Medpharm GmbH Scientific Publishers: Stuttgart, Germany, 2004. [Google Scholar]
- Khela, S. Valeriana Officinalis; The IUCN Red List of Threatened Species: Gland, Switzerland, 2012. [Google Scholar]
- Bell, C.D. Preliminary phylogeny of Valerianaceae (Dipsacales) inferred from nuclear and chloroplast DNA sequence data. Mol. Phylogenet. Evol. 2004, 31, 340–350. [Google Scholar] [CrossRef] [PubMed]
- Zaja̜c, M.; Zajac, A. Elementy Geograficzne Rodzimej Flory Polski: The Geographical Elements of Native Flora of Poland; Inst. Botaniki Uniw. Jagiellońskiego: Krakow, Poland, 2009; p. 93. [Google Scholar]
- Bone, K.; Simon Mills, M.; Fnimh, M. Principles and Practice of Phytotherapy: Modern Herbal Medicine; Elsevier Health Sciences: London, UK, 2012; p. 1056. [Google Scholar]
- Evstatieva, L.; Handjieva, N.; Popov, S.; Pashankov, P. A biosystematic study of Valeriana officinalis (Valerianaceae) distributed in Bulgaria. Plant Sys. Evol. 1993, 185, 167–179. [Google Scholar] [CrossRef]
- Hidalgo, O.; Vallès, J. First record of a natural hexaploid population for Valeriana officinalis: genome size is confirmed to be a suitable indicator of ploidy level in the species. Caryologia 2012, 65, 243–245. [Google Scholar] [CrossRef]
- Skalińska, M. Polyploidy in Valeriana officinalis Linn. in relation to its ecology and distribution. Bot. J. Linn. Soc. 1947, 53, 159–186. [Google Scholar] [CrossRef]
- Egan, A.N.; Schlueter, J.; Spooner, D.M. Applications of next-generation sequencing in plant biology. Am. J. Bot. 2012, 99, 175–185. [Google Scholar] [CrossRef] [Green Version]
- Andrews, K.R.; Good, J.M.; Miller, M.R.; Luikart, G.; Hohenlohe, P.A. Harnessing the power of RADseq for ecological and evolutionary genomics. Nat. Rev. Genet. 2016, 17, 81. [Google Scholar] [CrossRef] [Green Version]
- Davey, J.W.; Hohenlohe, P.A.; Etter, P.D.; Boone, J.Q.; Catchen, J.M.; Blaxter, M.L. Genome-wide genetic marker discovery and genotyping using next-generation sequencing. Nat. Rev. Genet. 2011, 12, 499. [Google Scholar] [CrossRef]
- Poland, J.A.; Brown, P.J.; Sorrells, M.E.; Jannink, J.-L. Development of high-density genetic maps for barley and wheat using a novel two-enzyme genotyping-by-sequencing approach. PloS ONE 2012, 7, e32253. [Google Scholar] [CrossRef] [Green Version]
- Jaccoud, D.; Peng, K.; Feinstein, D.; Kilian, A. Diversity arrays: A solid state technology for sequence information independent genotyping. Nucleic Acids Res. 2001, 29, 25. [Google Scholar] [CrossRef] [Green Version]
- Cruz, V.M.V.; Kilian, A.; Dierig, D.A. Development of DArT marker platforms and genetic diversity assessment of the US collection of the new oilseed crop Lesquerella and related species. PLoS ONE 2013, 8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rutherford, S.; Rossetto, M.; Bragg, J.G.; McPherson, H.; Benson, D.; Bonser, S.P.; Wilson, P.G. Speciation in the presence of gene flow: Population genomics of closely related and diverging Eucalyptus species. Heredity 2018, 121, 126–141. [Google Scholar] [CrossRef] [PubMed]
- Rucińska, A.; Polish Academy of Sciences Botanical Garden - Center for Biological Diversity Conservation in Powsin, Warsaw, Poland. Personal communication, 2018.
- Braun-Blanquet, J. Plant Sociology, The Study of Plant Communities; McGraw Hill: New York, NY, USA, 1932. [Google Scholar]
- NCPGR. EGISET. Available online: https://wyszukiwarka.ihar.edu.pl/pl (accessed on 1 July 2020).
- Jaccard, P. Étude comparative de la distribution florale dans une portion des Alpes et des Jura. Comparative study of the floral distribution in a part of the Alps and the Jura. Bull. Soc. Vaudoise. Sci. Nat. 1901, 37, 547–579. [Google Scholar]
- Evanno, G.; Regnaut, S.; Goudet, J. Detecting the number of clusters of individuals using the software STRUCTURE: A simulation study. Mol. Ecol. 2005, 14, 2611–2620. [Google Scholar] [CrossRef] [Green Version]
- Peakall, R.; Smouse, P.E. GenAlEx 6.5: Genetic analysis in Excel. Population genetic software for teaching and research-an update. Bioinformatics 2012, 28, 2537–2539. [Google Scholar] [CrossRef] [Green Version]
- Hubisz, M.J.; Falush, D.; Stephens, M.; Pritchard, J.K. Inferring weak population structure with the assistance of sample group information. Mol. Ecol. Res. 2009, 9, 1322–1332. [Google Scholar] [CrossRef] [Green Version]
- Kopelman, N.M.; Mayzel, J.; Jakobsson, M.; Rosenberg, N.A.; Mayrose, I. Clumpak: A program for identifying clustering modes and packaging population structure inferences across K. Mol. Ecol. Res. 2015, 15, 1179–1191. [Google Scholar] [CrossRef] [Green Version]
- Barton, D.L.; Atherton, P.J.; Bauer, B.A.; Moore, D.F., Jr.; Mattar, B.I.; LaVasseur, B.I.; Rowland, K.M., Jr.; Zon, R.T.; LeLindqwister, N.A.; Nagargoje, G.G. The use of Valeriana officinalis (valerian) in improving sleep in patients who are undergoing treatment for cancer: A phase III randomized, placebo-controlled, double-blind study: NCCTG Trial, N01C5. J. Support. Oncol. 2011, 9, 24. [Google Scholar] [CrossRef] [Green Version]
- Circosta, C.; De Pasquale, R.; Samperi, S.; Pino, A.; Occhiuto, F. Biological and analytical characterization of two extracts from Valeriana officinalis. J. Ethnopharmacol. 2007, 112, 361–367. [Google Scholar] [CrossRef]
- Letchamo, W.; Ward, W.; Heard, B.; Heard, D. Essential oil of Valeriana officinalis L. cultivars and their antimicrobial activity as influenced by harvesting time under commercial organic cultivation. J. Agr. Food Chem. 2004, 52, 3915–3919. [Google Scholar] [CrossRef]
- Singh, N.; Gupta, A.; Singh, B.; Kaul, V. Quantification of valerenic acid in Valeriana jatamansi and Valeriana officinalis by HPTLC. Chromatographia 2006, 63, 209–213. [Google Scholar] [CrossRef]
- Bressler, S.; Klatte-Asselmeyer, V.; Fischer, A.; Paule, J.; Dobeš, C. Variation in genome size in the Valeriana officinalis complex resulting from multiple chromosomal evolutionary processes. Preslia 2017, 89, 41–61. [Google Scholar] [CrossRef] [Green Version]
- Pant, M.; Nailwal, T.K.; Tewari, L.M.; Kumar, S.; Kumari, P.; Kholia, H.; Tewari, G.; Campus, D. Molecular characterization of Valeriana species with PCR, RAPD and SDS PAGE. Nat. Sci. 2009, 7, 41–49. [Google Scholar]
- Penzkofer, M.; Seefelder, S.; Heuberger, H. Estimation of outcrossing rates using genomic marker and determination of seed quality parameters in Valeriana officinalis L. sl under field conditions. Euphytica 2018, 214, 81. [Google Scholar] [CrossRef]
- Rajkumar, S.; Singh, S.K.; Nag, A.; Ahuja, P.S. Genetic Structure of Indian Valerian (Valeriana jatamansi) Populations in Western Himalaya Revealed by AFLP. Biochem. Genet. 2011, 49, 674–681. [Google Scholar] [CrossRef]
- Singh, S.K.; Katoch, R.; Kapila, R.K. Genetic and Biochemical Diversity among Valeriana jatamansi Populations from Himachal Pradesh. Sci. World J. 2015, 2015, 1–10. [Google Scholar]
- Sundaresan, V.; Sahni, G.; Verma, R.; Padalia, R.; Mehrotra, S.; Thul, S.T. Impact of geographic range on genetic and chemical diversity of Indian valerian (Valeriana jatamansi) from northwestern Himalaya. Biochem. Genet. 2012, 50, 797–808. [Google Scholar] [CrossRef]
- Jugran, A.; Rawat, S.; Dauthal, P.; Mondal, S.; Bhatt, I.D.; Rawal, R.S. Association of ISSR markers with some biochemical traits of Valeriana jatamansi Jones. Ind. Crop. Prod. 2013, 44, 671–676. [Google Scholar] [CrossRef]
- Jugran, A.K.; Bhatt, I.D.; Rawal, R.S.; Nandi, S.K.; Pande, V. Patterns of morphological and genetic diversity of Valeriana jatamansi Jones in different habitats and altitudinal range of West Himalaya, India. Flora 2013, 208, 13–21. [Google Scholar] [CrossRef]
- Jugran, A.K.; Bhatt, I.D.; Mondal, S.; Rawal, R.S.; Nandi, S.K. Genetic diversity assessment of Valeriana jatamansi Jones using microsatellites markers. Curr. Sci. 2015, 109, 1273–1282. [Google Scholar] [CrossRef] [Green Version]
- Gonzalez, L.A. Phylogenetics and mating system evolution in the Southern South American Valeriana (Valerianaceae). Master’s Thesis, University of New Orleans, New Orleans, LA, USA, 13 August 2014. [Google Scholar]
- Edet, O.U.; Gorafi, Y.S.; Nasuda, S.; Tsujimoto, H. DArTseq-based analysis of genomic relationships among species of tribe Triticeae. Sci. Rep. 2018, 8. [Google Scholar] [CrossRef]
- Zaitoun, S.Y.A.; Jamous, R.M.; Shtaya, M.J.; Mallah, O.B.; Eid, I.S.; Ali-Shtayeh, M.S. Characterizing Palestinian snake melon (Cucumis melo var. flexuosus) germplasm diversity and structure using SNP and DArTseq markers. BMC Plant Biol. 2018, 18, 246. [Google Scholar]
- Alam, M.; Neal, J.; O’Connor, K.; Kilian, A.; Topp, B. Ultra-high-throughput DArTseq-based silicoDArT and SNP markers for genomic studies in macadamia. PloS ONE 2018, 13, e0203465. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schultz, A.J.; Cristescu, R.H.; Littleford-Colquhoun, B.L.; Jaccoud, D.; Frère, C.H. Fresh is best: Accurate SNP genotyping from koala scats. Ecol. Evol. 2018, 8, 3139–3151. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, N.H.; Premachandra, H.; Kilian, A.; Knibb, W. Genomic prediction using DArT-Seq technology for yellowtail kingfish Seriola lalandi. BMC Genom. 2018, 19, 107. [Google Scholar] [CrossRef]
- Melville, J.; Haines, M.L.; Boysen, K.; Hodkinson, L.; Kilian, A.; Smith Date, K.L.; Potvin, D.A.; Parris, K.M. Identifying hybridization and admixture using SNPs: Application of the DArTseq platform in phylogeographic research on vertebrates. R. Soc. Open Sci. 2017, 4, 161061. [Google Scholar] [CrossRef] [Green Version]
- Baloch, F.S.; Alsaleh, A.; Shahid, M.Q.; Çiftçi, V.; de Miera, L.E.S.; Aasim, M.; Nadeem, M.A.; Aktaş, H.; Özkan, H.; Hatipoğlu, R. A whole genome DArTseq and SNP analysis for genetic diversity assessment in durum wheat from central fertile crescent. PLoS ONE 2017, 12, e0167821. [Google Scholar] [CrossRef] [Green Version]
- Al-Beyroutiová, M.; Sabo, M.; Sleziak, P.; Dušinský, R.; Birčák, E.; Hauptvogel, P.; Kilian, A.; Švec, M. Evolutionary relationships in the genus Secale revealed by DArTseq DNA polymorphism. Plant Syst. Evol. 2016, 302, 1083–1091. [Google Scholar] [CrossRef]
- Targońska-Karasek, M.; Bolibok-Brągoszewska, H.; Rakoczy-Trojanowska, M. DArTseq genotyping reveals high genetic diversity of polish rye inbred lines. Crop Sci. 2017, 57, 1906–1915. [Google Scholar] [CrossRef] [Green Version]
- Bauer, E.; Schmutzer, T.; Barilar, I.; Mascher, M.; Gundlach, H.; Martis, M.M.; Twardziok, S.O.; Hackauf, B.; Gordillo, A.; Wilde, P. Towards a whole-genome sequence for rye (Secale cereale L.). Plant J. 2017, 89, 853–869. [Google Scholar] [CrossRef] [Green Version]
- Bennett, M.D.; Leitch, I.J. Nuclear DNA amounts in angiosperms. Ann. Bot. 1995, 76, 113–176. [Google Scholar] [CrossRef]
- Kostrakiewicz-Gierałt, K. The variability of population and individual traits of medicinal plant Valeriana officinalis L. var. officinalis Mikan under different site conditions. Period. Biol. 2018, 120, 41–50. [Google Scholar]
- Konon, N.; Novikova, N. Reaction of Valeriana officinalis to inbreeding. Rastitel’nye Resur. 1981, 17, 85–90. [Google Scholar]
- Ruosteenoja, K.; Markkanen, T.; Venäläinen, A.; Räisänen, P.; Peltola, H. Seasonal soil moisture and drought occurrence in Europe in CMIP5 projections for the 21st century. Clim. Dyn. 2018, 50, 1177–1192. [Google Scholar] [CrossRef] [Green Version]
- Brook, B.W.; Tonkyn, D.W.; O’Grady, J.J.; Frankham, R. Contribution of inbreeding to extinction risk in threatened species. Conserv. Ecol. 2002, 6, 1–16. [Google Scholar] [CrossRef] [Green Version]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Population No. | Region—Voivodeship | Accession No. | Latitude | Longitude | Abundance | Ploidy |
|---|---|---|---|---|---|---|
| 1 | Kujawsko-Pomorskie | PL 401930 | N 53.03 | E 17.11 | 4 | 2n = 14 |
| 2 | Podkarpackie | PL 401935 | N 49.26 | E 22.03 | 2 | 2n = 14 |
| 3 | Podkarpackie | PL 403179 | N 49.21 | E 22.09 | 3 | 2n = 14 |
| 4 | Podkarpackie | PL 403181 | N 49.25 | E 22.07 | 2 | 2n = 14 |
| 5 | Podkarpackie | PL 403182 | N 49.39 | E 21.59 | 2 | 2n = 14 |
| 6 | Podkarpackie | PL 403183 | N 49.36 | E 22.12 | 1 | 2n = 14 |
| 7 | Podkarpackie | PL 403184 | N 49.41 | E 22.15 | 1 | 2n = 14 |
| 8 | Świętokrzyskie | PL 403186 | N 50.43 | E 20.31 | 2 | 2n = 14 |
| 9 | Świętokrzyskie | PL 403187 | N 50.43 | E 20.31 | 4 | 2n = 14 |
| 10 | Świętokrzyskie | PL 401938 | N 50.46 | E 20.32 | 3 | 2n = 14 |
| 11 | Świętokrzyskie | PL 401939 | N 50.52 | E 20.56 | 3 | 2n = 14 |
| 12 | Świętokrzyskie | PL 401941 | N 50.92 | E 20.56 | 2 | 2n = 14 |
| 13 | Świętokrzyskie | PL 401942 | N 51.01 | E 20.26 | 2 | 2n = 14 |
| 14 | Świętokrzyskie | PL 403190 | N 51.01 | E 20.28 | 3 | 2n = 14 |
| 15 | Świętokrzyskie | PL 401945 | N 51.04 | E 20.23 | 4 | 2n = 14 |
| 16 | Świętokrzyskie | PL 401946 | N 51.14 | E 20.22 | 3 | 2n = 14 |
| 17 | Świętokrzyskie | PL 403191 | N 51.06 | E 20.15 | 3 | 2n = 14 |
| 18 | Mazowieckie | PL 406738 | N 52.07 | E 21.05 | 2 | 2n = 14 |
| 19 | Mazowieckie | PL 403192 | N 52.07 | E 21.05 | 2 | 2n = 14 |
| 20 | Cultivar “Lubelski” | PL 401951 | N 52.81 | E 20.18 | – | 2n = 28 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Boczkowska, M.; Bączek, K.; Kosakowska, O.; Rucińska, A.; Podyma, W.; Węglarz, Z. Genome-Wide Diversity Analysis of Valeriana officinalis L. Using DArT-seq Derived SNP Markers. Agronomy 2020, 10, 1346. https://doi.org/10.3390/agronomy10091346
Boczkowska M, Bączek K, Kosakowska O, Rucińska A, Podyma W, Węglarz Z. Genome-Wide Diversity Analysis of Valeriana officinalis L. Using DArT-seq Derived SNP Markers. Agronomy. 2020; 10(9):1346. https://doi.org/10.3390/agronomy10091346
Chicago/Turabian StyleBoczkowska, Maja, Katarzyna Bączek, Olga Kosakowska, Anna Rucińska, Wiesław Podyma, and Zenon Węglarz. 2020. "Genome-Wide Diversity Analysis of Valeriana officinalis L. Using DArT-seq Derived SNP Markers" Agronomy 10, no. 9: 1346. https://doi.org/10.3390/agronomy10091346