
Synthesis of Hyperbranched Poly(ε-caprolactone) Containing Terminal Azobenzene Structure via Combined Ring-Opening Polymerization and “Click” Chemistry

Abstract
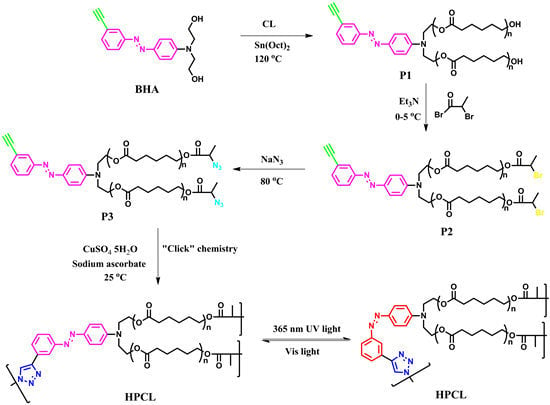
:
1. Introduction
2. Experimental Section
2.1. Materials
2.2. Analysis and Characterizations
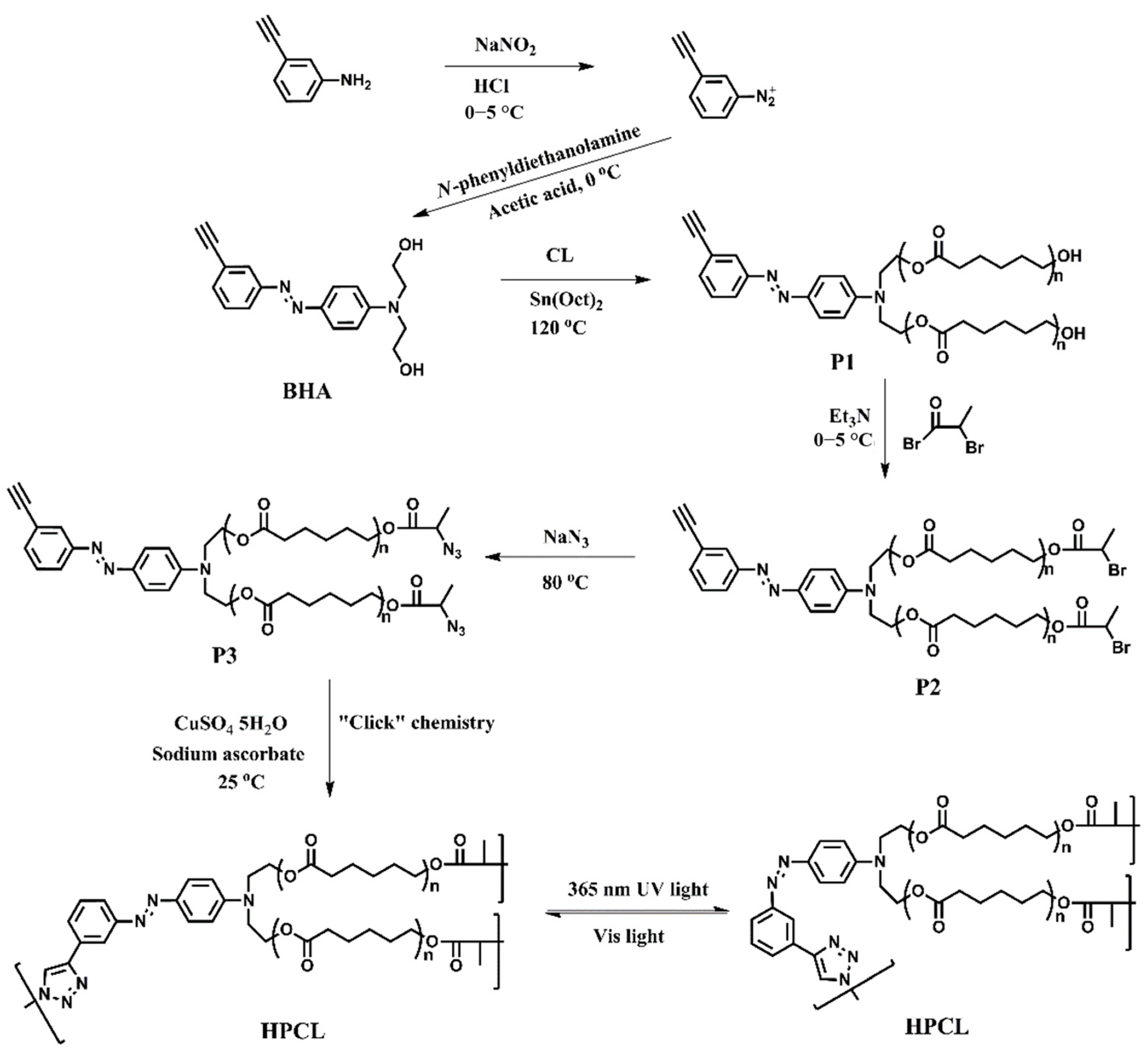
2.3. Synthesis of N,N′-Bis(2-hydroxyethyl)-4-(3-ethynylphenylazo)aniline (BHA)
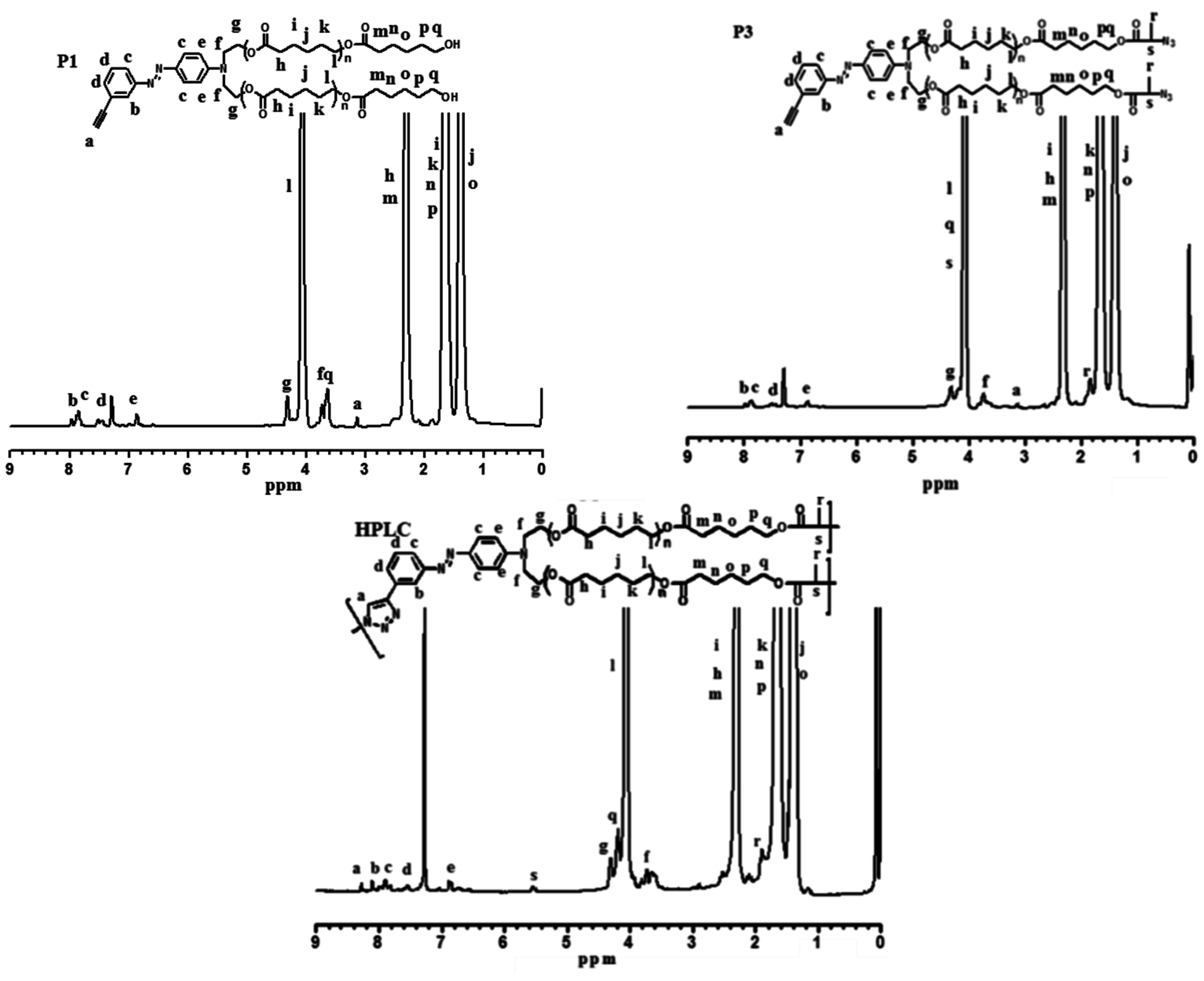
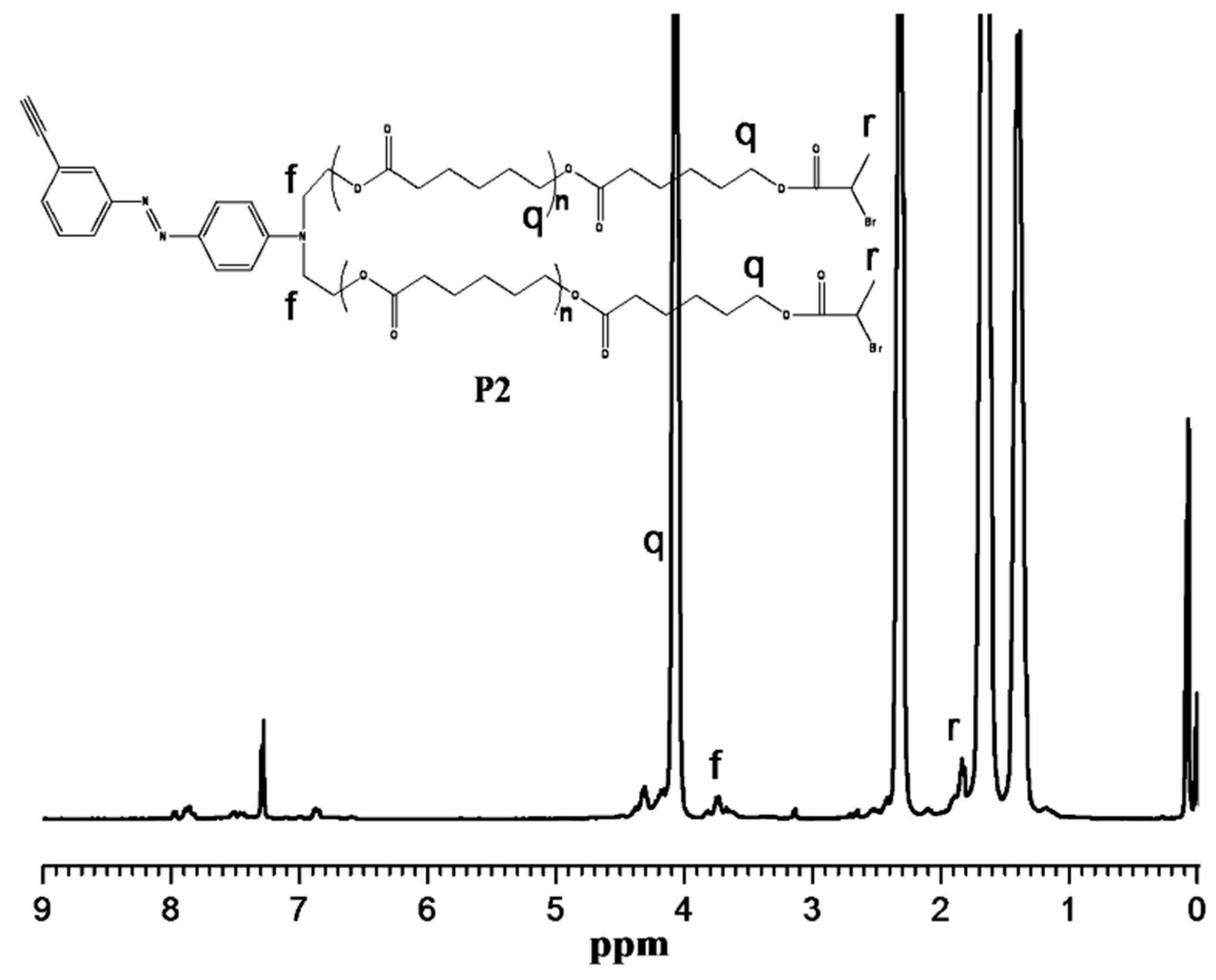
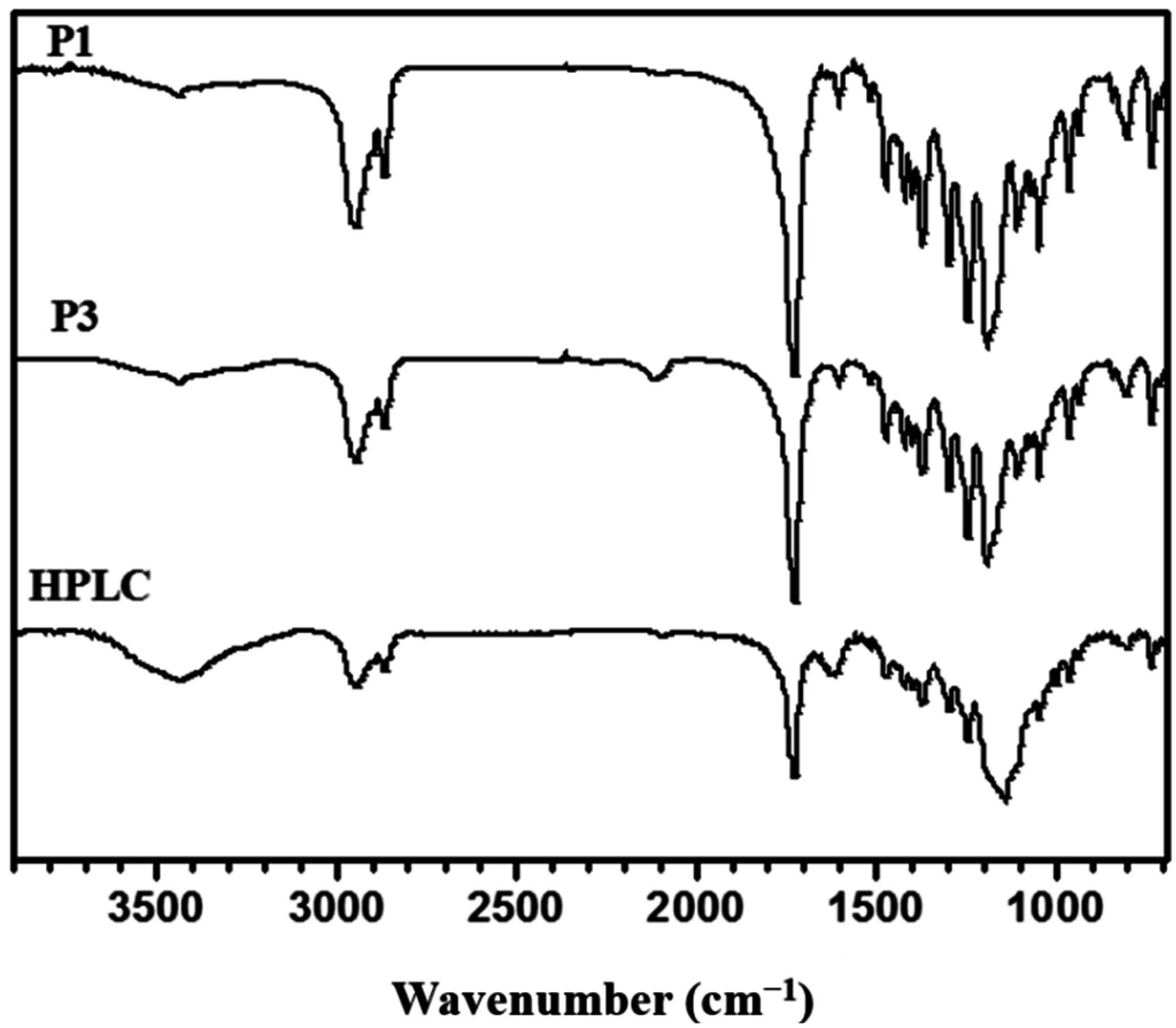
2.4. Synthesis of Poly(ε-caprolactone) Containing Terminal Azobenzene and Ethyne Groups (P1)
2.5. Synthesis of AB2 Type Macromonomer (P3)

2.6. Synthesis of Hyperbranched Poly(ε-caprolactone) (HPCL)
3. Results and Discussion
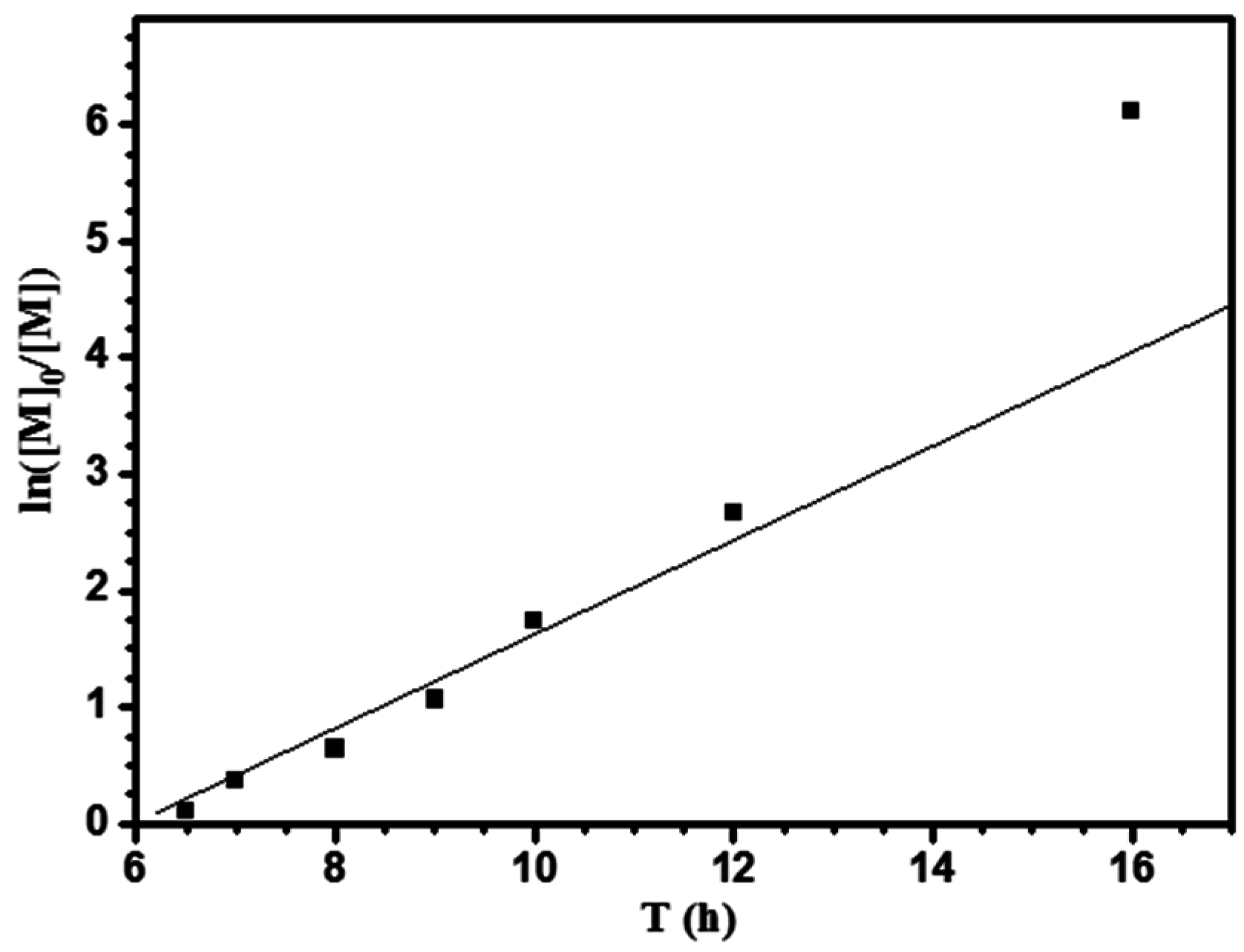
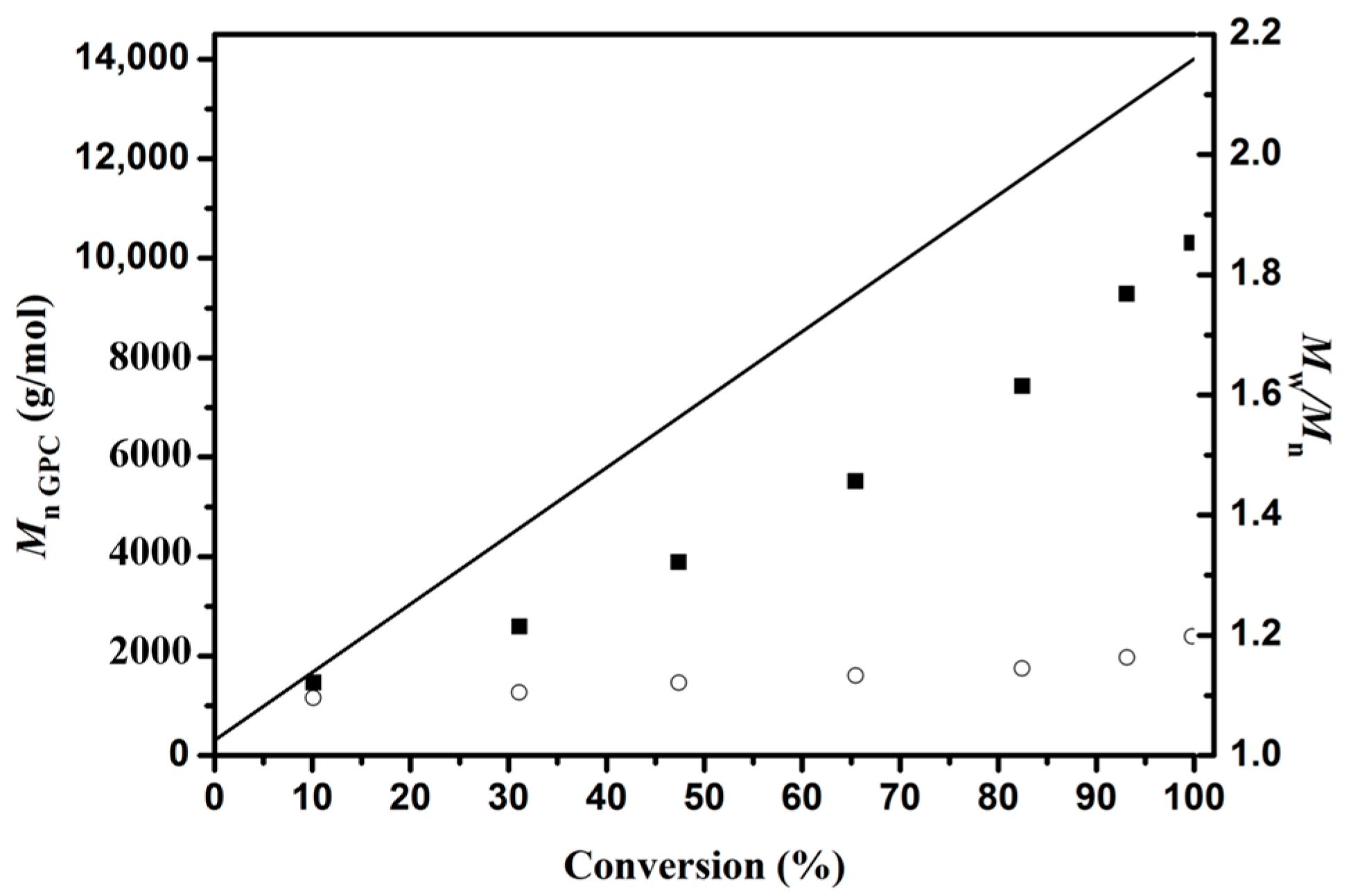
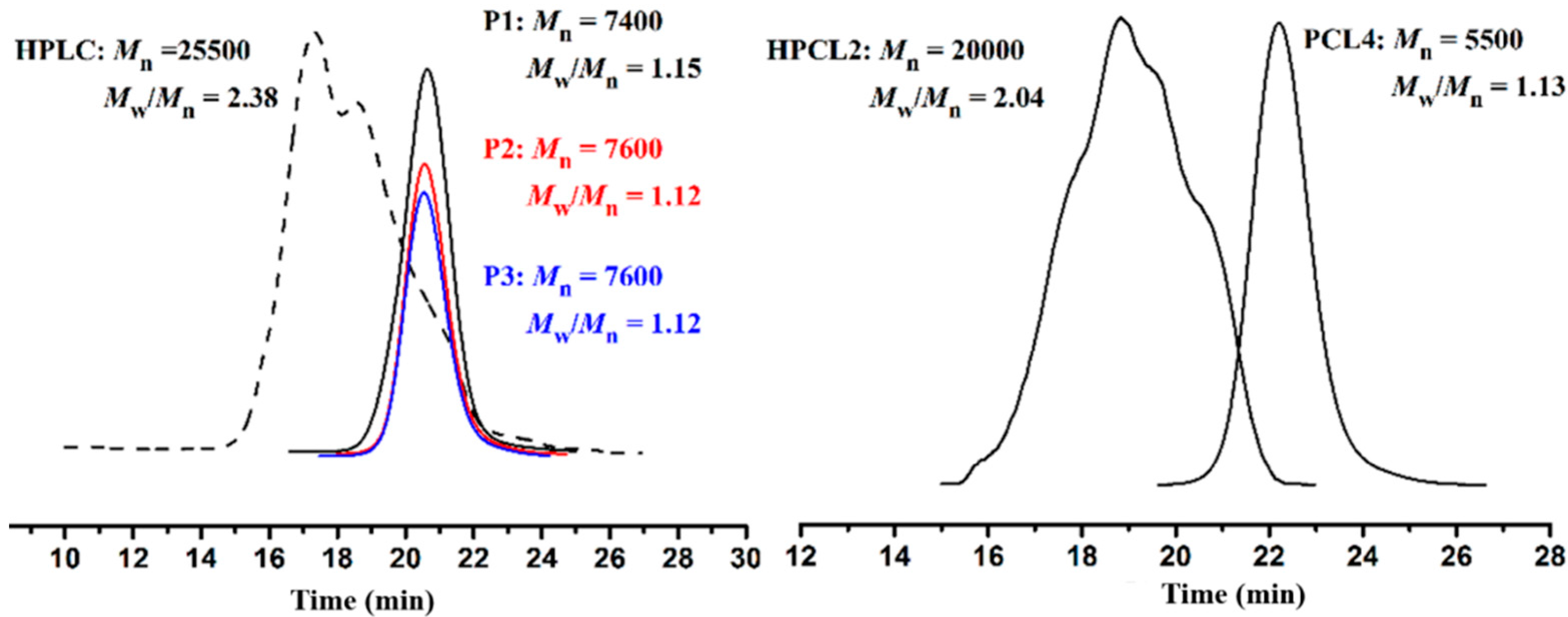
3.1. Ring-opening Polymerization of ε-Caprolactone (CL)



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample a | Time b (h) | Conversion c (%) | Mn GPC d (g/mol) | Mn the g/mol | Mw/Mn f | Mn NMR g (g/mol) |
|---|---|---|---|---|---|---|
| PCL1 | 6.5 | 10.10 | 1,400 | 1,700 | 1.10 | 1,500 |
| PCL2 | 7 | 31.11 | 2,600 | 4,500 | 1.10 | 4,200 |
| PCL3 | 8 | 47.37 | 3,900 | 6,800 | 1.12 | 6,600 |
| PCL4 | 9 | 65.44 | 5,500 | 9,300 | 1.13 | 8,600 |
| P1 | 10 | 82.45 | 7,400 | 11,600 | 1.14 | 11,300 |
| PCL5 | 12 | 93.12 | 9,300 | 13,100 | 1.16 | 13,200 |
| PCL6 | 16 | 99.78 | 10,300 | 14,000 | 1.20 | 13,500 |
3.2. “Click” Chemistry for Hyperbranched Poly(ε-caprolactone) (HPCL)




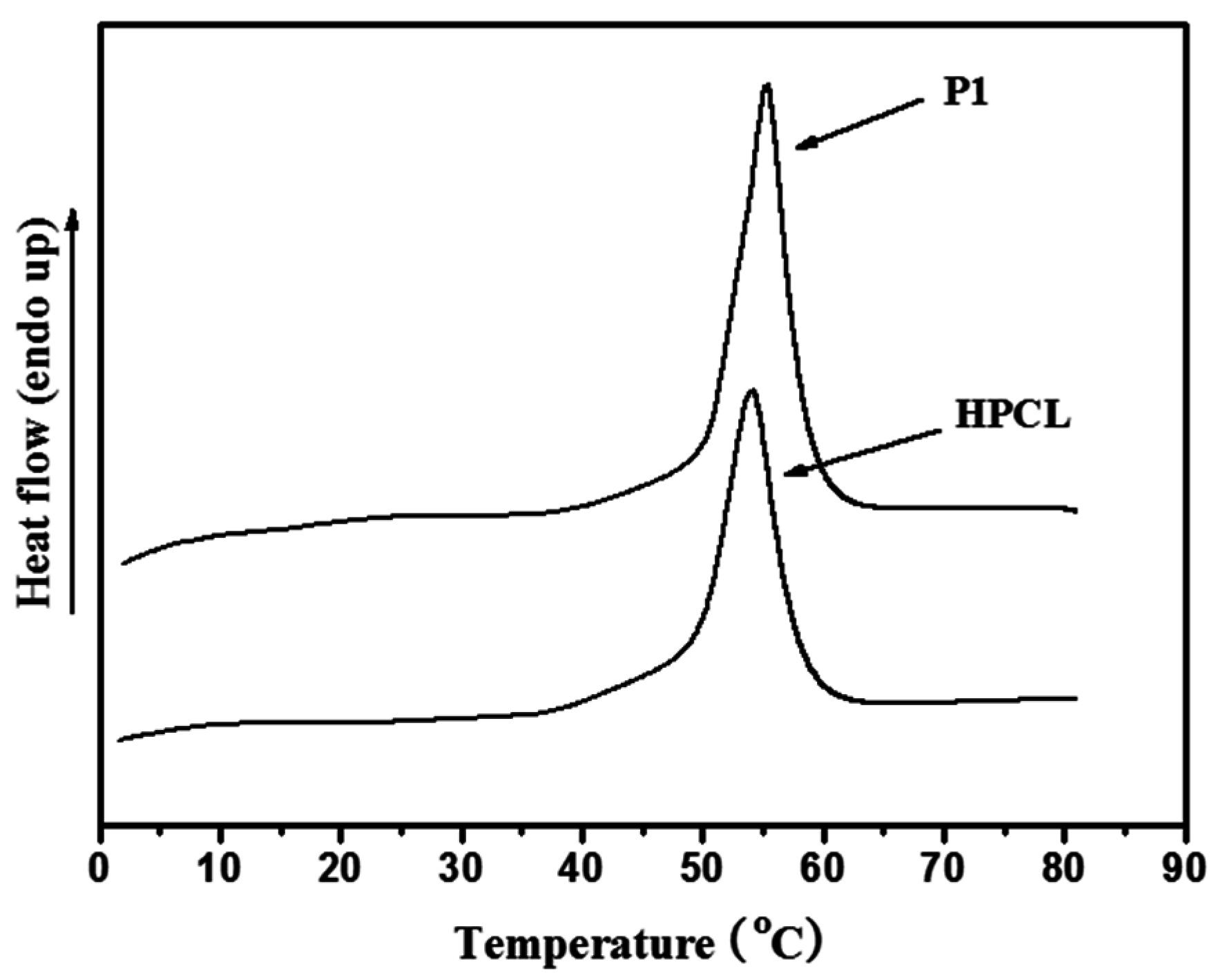
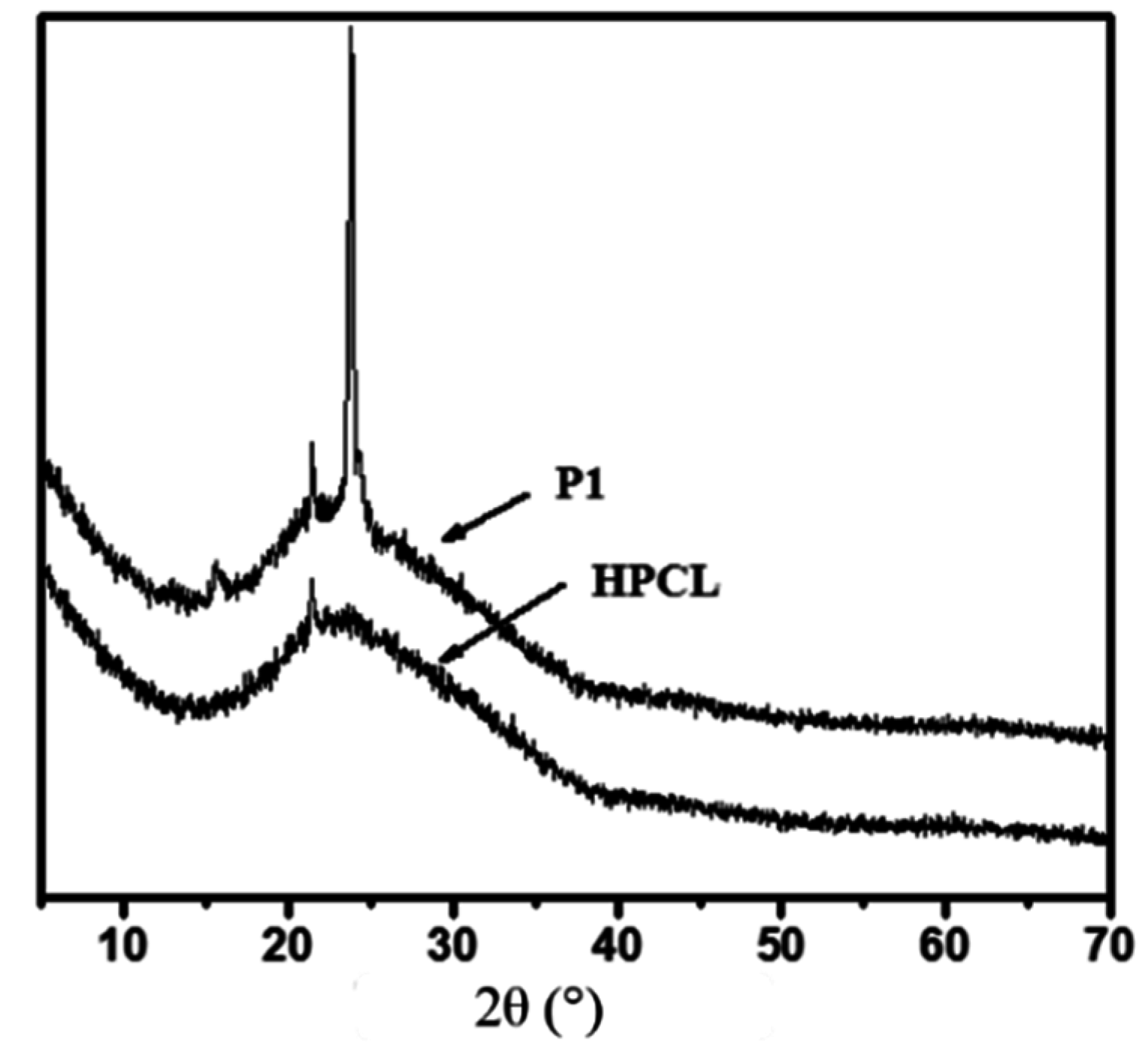
3.3. Thermal and Crystallization Characterization



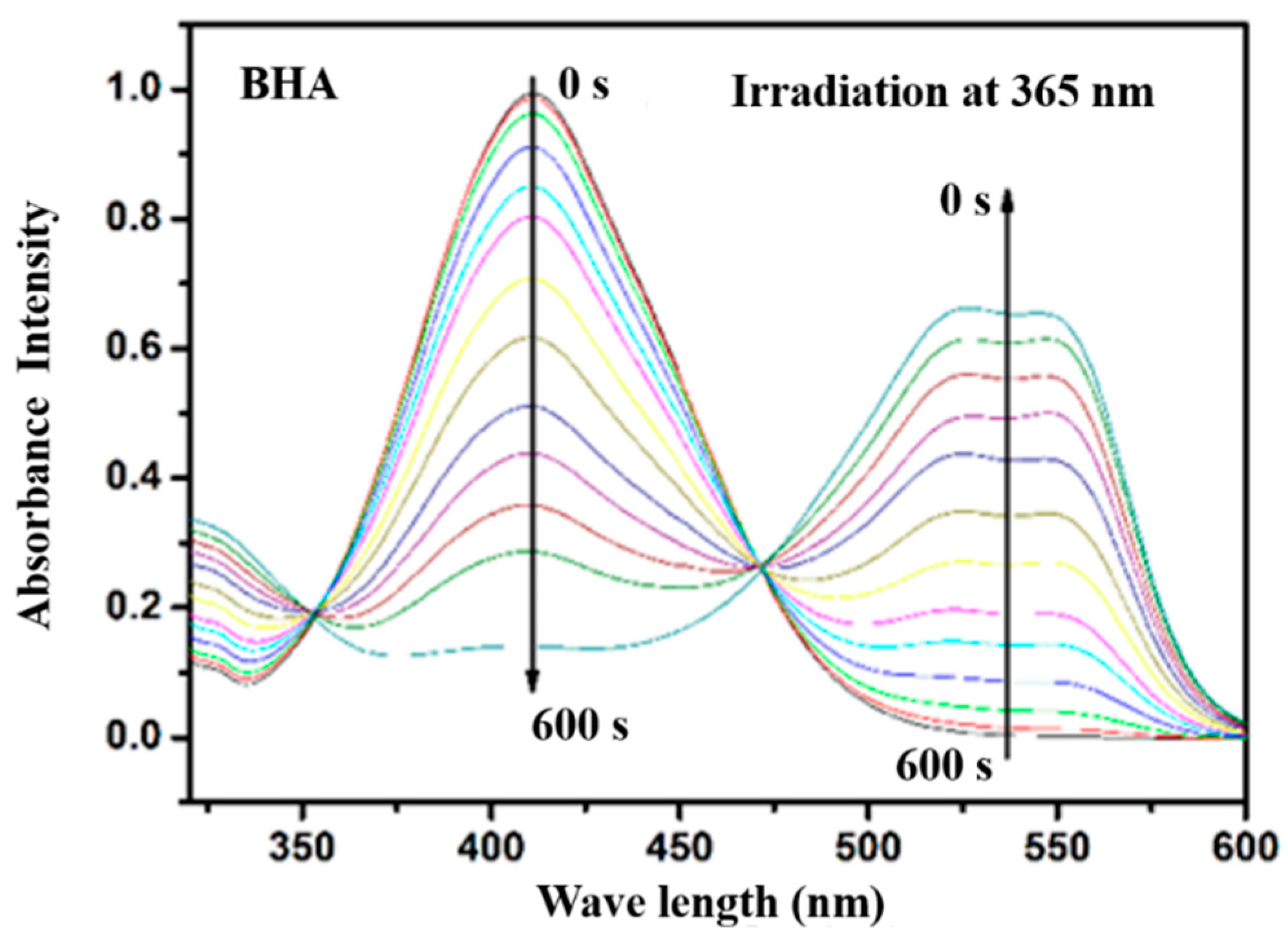
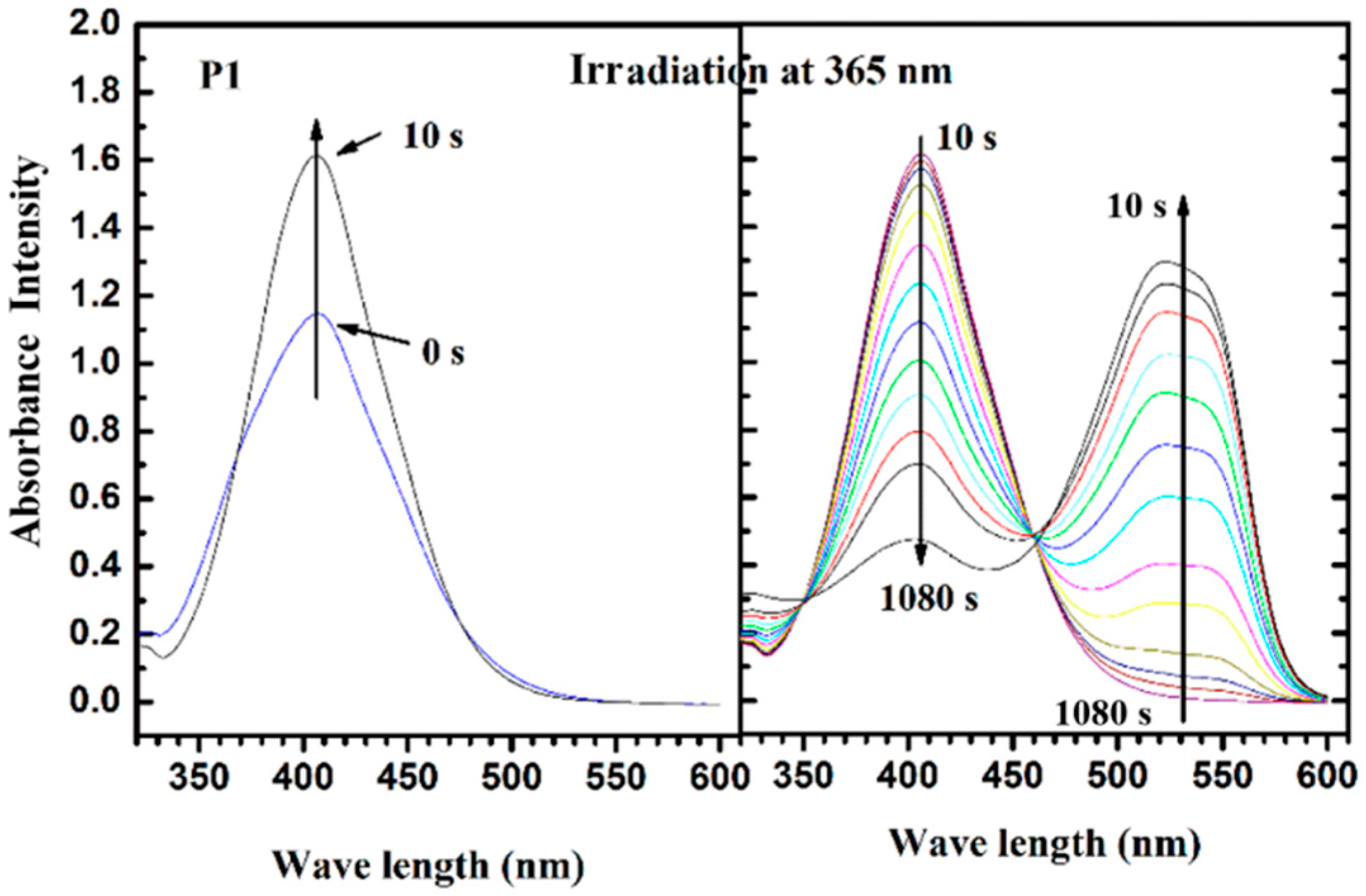
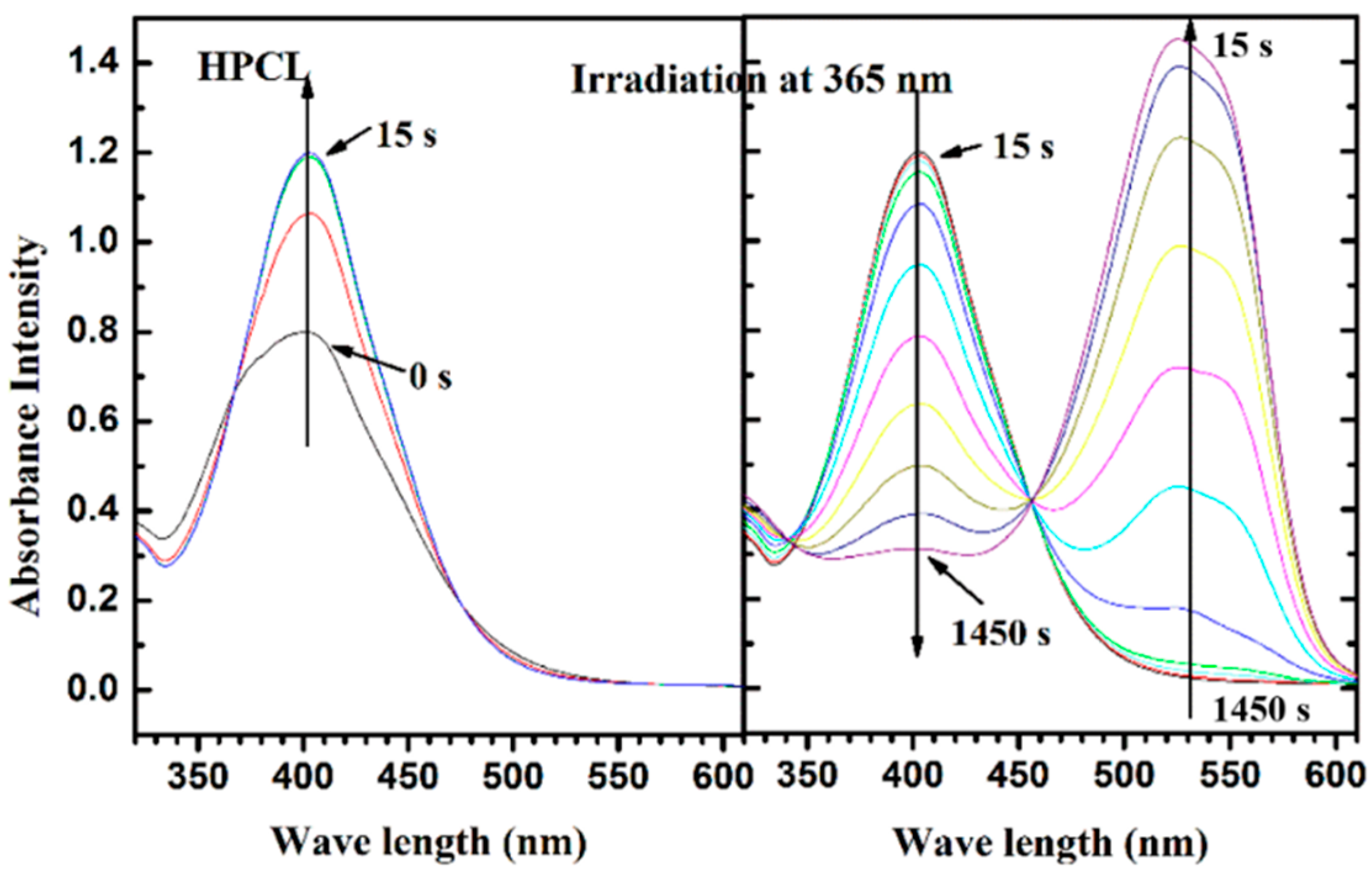
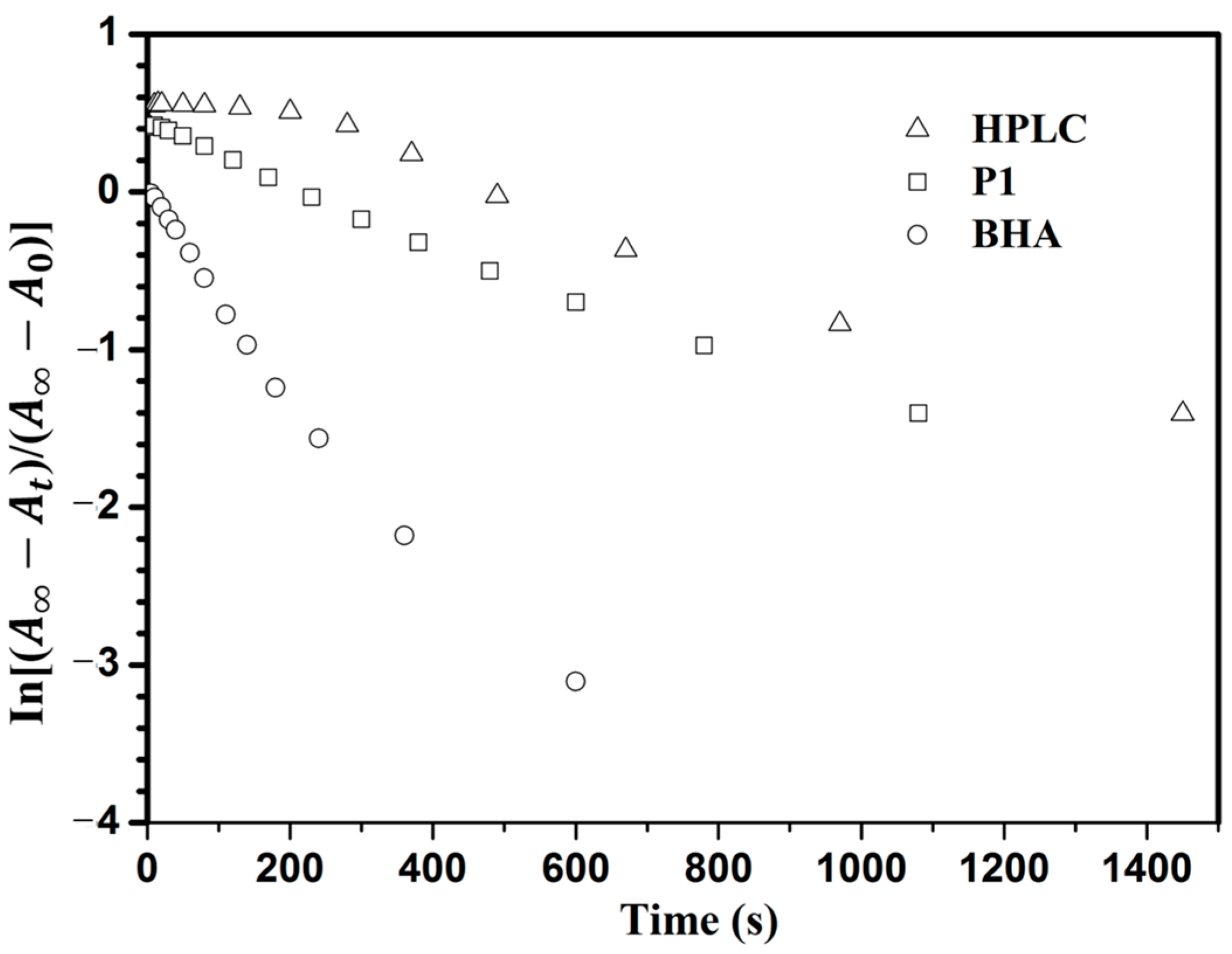
3.4. Photoisomerization Behaviors





4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Albertsson, A.C.; Varma, I.K. Recent developments in ring opening polymerization of lactones for biomedical applications. Biomacromolecules 2003, 4, 1466–1486. [Google Scholar] [CrossRef] [PubMed]
- Holland, S.J.; Tighe, B.J.; Gould, P.L.J. Polymers for biodegradable medical devices. 1. The potential of polyesters as controlled macromolecular release systems. Control. Release 1986, 4, 155–180. [Google Scholar] [CrossRef]
- Pitt, C.G. Poly-ε-caprolactone and Its Copolymers; Chasin, M., Langer, R., Eds.; Marcel Dekker: New York, NY, USA, 1990; Volume 45, pp. 71–120. [Google Scholar]
- Eastmond, G.C. Poly(ε-caprolactone) blends. In Biomedical Applications Polymer Blends; Eastmond, G.C., Höcker, H., Klee, D., Eds.; Springer: Berlin/Heidelberg, Germany, 1999; Volume 149, pp. 59–223. [Google Scholar]
- Benedict, C.V.; Cameron, J.A.; Huang, S.J. Polycaprolactone degradation by mixed and pure cultures of bacteria and a yeast. J. Appl. Polym. Sci. 1983, 28, 335–342. [Google Scholar] [CrossRef]
- Lang, M.D.; Wong, R.P.; Chu, C.C. Synthesis and structural analysis of functionalized poly(ε-caprolactone)-based three-arm star polymers. J. Polym. Sci. Part A 2002, 40, 1127–1141. [Google Scholar] [CrossRef]
- An, S.G.; Cho, C.G. Synthesis and characterization of amphiphilic poly(caprolactone) star block copolymers. Macromol. Rapid Commun. 2004, 25, 618–622. [Google Scholar] [CrossRef]
- Bahadori, F.; Dag, A.; Durmaz, H.; Cakir, N.; Onyuksel, H.; Tunca, U.; Topcu, G.; Hizal, G. Synthesis and characterization of biodegradable amphiphilic star and Y-shaped block copolymers as potential carriers for vinorelbine. Polymers 2014, 6, 214–242. [Google Scholar] [CrossRef]
- Nunez, E.; Ferrando, C.; Malmstrom, E.; Claesson, H.; Werner, P.E.; Gedde, U.W. Crystal structure, melting behaviour and equilibrium melting point of star polyesters with crystallisable poly(ε-caprolactone) arms. Polymer 2004, 45, 5251–5263. [Google Scholar] [CrossRef]
- Kwak, S.Y.; Choi, J.; Song, H.J. Viscoelastic relaxation and molecular mobility of hyperbranched poly(ε-caprolactone)s in their melt state. Chem. Mater. 2005, 17, 1148–1156. [Google Scholar] [CrossRef]
- Flory, P.J. Molecular size distribution in three dimensional polymers. 6 branched polymers containing A-R-BF-1 type units. J. Am. Chem. Soc. 1952, 74, 2718–2723. [Google Scholar] [CrossRef]
- Kim, Y.H.; Webster, O.W. Water soluble hyperbranched polyphenylene: “A unimolecular micelle?”. J. Am. Chem. Soc. 1990, 112, 4592–4593. [Google Scholar] [CrossRef]
- Jiang, L.; Huang, W.Y.; Xue, X.Q.; Yang, H.J.; Jiang, B.B.; Zhang, D.L.; Fang, J.B.; Chen, J.H.; Yang, Y.; Zhai, G.Q.; et al. Radical polymerization in the presence of chain transfer monomer: An approach to branched vinyl polymers. Macromolecules 2012, 45, 4092–4100. [Google Scholar] [CrossRef]
- Yang, H.J.; Jiang, B.B.; Huang, W.Y.; Zhang, D.L.; Kong, L.Z.; Chen, J.; Liu, C.; Gong, F.H.; Yu, Q.; Yang, Y. Development of branching in atom transfer radical copolymerization of styrene with triethylene glycol dimethacrylate. Macromolecules 2009, 42, 5976–5982. [Google Scholar] [CrossRef]
- Xue, X.Q.; Li, F.; Huang, W.Y.; Yang, H.J.; Jiang, B.B.; Zheng, Y.L.; Zhang, D.L.; Fang, J.B.; Kong, L.Z.; Zhai, G.Q.; et al. Quadrangular prism: A unique self-assembly from amphiphilic hyperbranched PMA-b-PAA. Macromol. Rapid Commun. 2014, 35, 330–336. [Google Scholar] [CrossRef] [PubMed]
- Van Genderen, M.H.P.; de Brabander-van den Berg, E.M.M.; Meijer, E.W. Poly(propylene imine) dendrimers. In Advances in Dendritic Molecules; Newkome, G.R., Ed.; JAI Press: Greenwich, CT, USA, 1994; Volume 1, pp. 61–210. [Google Scholar]
- Denkewalter, R.G.; Kolc, J.; Lukasavage, W.J. Macromolecular highly branched homogeneous compound based on lysine units. U.S. Patent 4289872, 15 September 1981. [Google Scholar]
- Jiang, Q.M.; Huang, W.Y.; Yang, H.J.; Xue, X.Q.; Jiang, B.B.; Zhang, D.L.; Fang, J.B.; Chen, J.H.; Yang, Y.; Zhai, G.Q.; et al. Radical emulsion polymerization with chain transfer monomer: An approach to branched vinyl polymers with high molecular weight and relatively narrow polydispersity. Polym. Chem. 2013, 6, 1863–1873. [Google Scholar] [CrossRef]
- Trollsas, M.; Hedrick, J.L. Hyperbranched poly(ε-caprolactone) derived from intrinsically branched AB2 macromonomers. Macromolecules 1998, 31, 4390–4395. [Google Scholar] [CrossRef]
- Trollsas, M.; Atthoff, B.; Claesson, H.; Hedrick, J.L. Hyperbranched poly(ε-caprolactone)s. Macromolecules 1998, 31, 3439–3445. [Google Scholar] [CrossRef]
- Liu, W.; Dong, C.M. Versatile strategy for the synthesis of hyperbranched poly(ε-caprolactone)s and polypseudorotaxanes thereof. Macromolecules 2010, 43, 8447–8455. [Google Scholar] [CrossRef]
- Choi, J.; Kwak, S.Y. Synthesis and characterization of hyperbranched poly(ε-caprolactone)s having different lengths of homologous backbone segments. Macromolecules 2003, 36, 8630–8637. [Google Scholar] [CrossRef]
- Choi, J.; Kwak, S.Y. Architectural effects of poly(ε-caprolactone)s on the crystallization kinetics. Macromolecules 2004, 37, 3745–3754. [Google Scholar] [CrossRef]
- Kolb, H.C.; Finn, M.G.; Sharpless, K.B. Click chemistry: Diverse chemical function from a few good reactions. Angew. Chem. Int. Ed. 2001, 40, 2004–2021. [Google Scholar] [CrossRef]
- Wang, Q.; Chan, T.R.; Hilgraf, R.; Fokin, V.V.; Sharpless, K.B.; Finn, M.G. Bioconjugation by copper(I)-catalyzed azide-alkyne [3+2] cycloaddition. J. Am. Chem. Soc. 2003, 125, 3192–3193. [Google Scholar] [CrossRef] [PubMed]
- Wu, P.; Feldman Alina, K.; Nugent Anne, K.; Hawker Craig, J.; Scheel, A.; Voit, B.; Pyun, J.; Frechet Jean, M.J.; Sharpless, K.B.; Fokin Valery, V. Efficiency and fidelity in a click-chemistry route to triazole dendrimers by the copper(I)-catalyzed ligation of azides and alkynes. Angew. Chem. Int. Ed. 2004, 43, 3928–3932. [Google Scholar] [CrossRef] [PubMed]
- Malkoch, M.; Schleicher, K.; Drockenmuller, E.; Hawker, C.J.; Russell, T.P.; Wu, P.; Fokin, V.V. Structurally diverse dendritic libraries: A highly efficient functionalization approach using click chemistry. Macromolecules 2005, 38, 3663–3678. [Google Scholar] [CrossRef]
- Elchinger, P.H.; Faugeras, P.A.; Boëns, B.; Brouillette, F.; Montplaisir, D.; Zerrouki, R.; Lucas, R. Polysaccharides: The “click” chemistry impact. Polymers 2011, 3, 1607–1651. [Google Scholar] [CrossRef]
- Xue, X.Q.; Zhu, J.; Zhang, Z.B.; Cheng, Z.P.; Tu, Y.F.; Zhu, X.L. Synthesis and characterization of azobenzene-functionalized poly(styrene)-b-poly(vinyl acetate) via the combination of RAFT and “click” chemistry. Polymer 2010, 51, 3083–3090. [Google Scholar] [CrossRef]
- Rudolph, T.; Crotty, S.; Lühe, M.V.D.; Pretzel, D.; Schubert, U.S.; Schacher, F.H. Thymine- and adenine-functionalized polystyrene form self-assembled structures through multiple complementary hydrogen bonds. Polymers 2013, 5, 1081–1101. [Google Scholar] [CrossRef]
- Van Hest, J.C.M.; Opsteen, J.A. Modular synthesis of block copolymers via cycloaddition of terminal azide and alkyne functionalized polymers. Chem. Commun. 2005, 57–59. [Google Scholar]
- Gao, H.F.; Matyjaszewski, K. Synthesis of star polymers by a combination of ATRP and the “click” coupling method. Macromolecules 2006, 39, 4960–4965. [Google Scholar] [CrossRef]
- Urbani, C.N.; Bell, C.A.; Whittaker, M.R.; Monteiro, M.J. Convergent synthesis of second generation AB-type miktoarm dendrimers using “click” chemistry catalyzed by copper wire. Macromolecules 2008, 41, 1057–1060. [Google Scholar] [CrossRef]
- Lin, W.C.; Fu, Q.; Zhang, Y.; Huang, J.L. One-pot synthesis of ABC type triblock copolymers via a combination of “click chemistry” and atom transfer nitroxide radical coupling chemistry. Macromolecules 2008, 41, 4127–4135. [Google Scholar] [CrossRef]
- Li, Z.A.; Hu, P.; Ye, C.; Liu, Y.; Qin, J.G.; Li, Z. New azo-chromophore-containing hyperbranched polytriazoles derived from AB2 monomers via click chemistry under copper(I) catalysis. Macromolecules 2009, 42, 1589–1596. [Google Scholar] [CrossRef]
- Qin, A.J.; Lam, J.W.Y.; Jim, C.K.W.; Zhang, L.; Yan, J.J.; Hussler, M.; Liu, J.Z.; Dong, Y.Q.; Liang, D.H.; Chen, E.Q.; et al. Hyperbranched polytriazoles: Click polymerization, regioisomeric structure, light emission, and fluorescent patterning. Macromolecules 2008, 41, 3808–3822. [Google Scholar] [CrossRef]
- Kronek, J.; Luston, J.; Bohme, F.; Komber, H. Azo-group labelled polyesters by end-capping with 2-oxazoline derivatives-preparation. Macromol. Symp. 2001, 164, 105–115. [Google Scholar]
- Kronek, J.; Luston, J.; Bohme, F.; Komber, H. Azo-group labelled polyesters by end-capping with 2-oxazoline derivatives-photochemical properties. Macromol. Symp. 2001, 170, 301–310. [Google Scholar] [CrossRef]
- Natansohn, A.; Rochon, P. Photoinduced motions in azo-containing polymers. Chem. Rev. 2002, 102, 4139–4175. [Google Scholar] [CrossRef] [PubMed]
- Kumar, G.; Neckers, D.C. Photochemistry of azobenzene-containing polymers. Chem. Rev. 1989, 89, 1915–1937. [Google Scholar] [CrossRef]
- Emoto, A.; Uchida, E.; Fukuda, T. Optical and physical applications of photocontrollable materials: Azobenzene-containing and liquid crystalline polymers. Polymers 2012, 4, 150–186. [Google Scholar] [CrossRef]
- Peptu, C.; Bogdan, V.H.; Simionescu, B.C.; Adamus, G.; Kowalczuk, M.; Nunzi, J.M. Disperse red 1 end capped oligoesters. Synthesis by noncatalyzed ring opening oligomerization and structural characterization. J. Polym. Sci. Part A 2009, 47, 534–547. [Google Scholar] [CrossRef]
- Wang, G.; Zhu, X.L.; Cheng, Z.P.; Zhu, J. Azobenzene-based initiator for atom transfer radical polymerization of methyl methacrylate. J. Polym. Sci. Part A 2005, 43, 2358–2367. [Google Scholar] [CrossRef]
- Wan, X.M.; Zhu, X.L.; Zhu, J.; Zhang, Z.B.; Cheng, Z.P. Synthesis of dithiocarbamate bearing azobenzene group and use for RAFT polymerization of vinyl monomers. J. Polym. Sci. Part A 2007, 45, 2886–2896. [Google Scholar] [CrossRef]
- Xue, X.Q.; Zhang, W.; Cheng, Z.P.; Zhu, J.; Zhu, X.L. A novel azo-containing dithiocarbamate used for living radical polymerization of methyl acrylate and styrene. J. Polym. Sci. Part A 2008, 46, 5626–5637. [Google Scholar] [CrossRef]
- Lee, J.H.; Choi, D.; Shin, E.J. Trans-cis isomerization of arylether dendrimers with azobenzene core and terminal hydroxy groups. Spectrochim. Acta Part A 2010, 77, 478–484. [Google Scholar] [CrossRef] [PubMed]
- Yu, L.P.; Zhu, J.; Cheng, Z.P.; Zhang, Z.B.; Zhang, W.; Zhu, X.L. Synthesis of azobenzene-centered (co)polymers via reversible addition-fragmentation chain transfer (RAFT) polymerization. e-Polymers 2013, 7, 5440–5448. [Google Scholar]
- Dubois, Ph.; Ropson, N.; Jerome, R.; Teyssie, P. Macromolecular engineering of polylactones and polylactides. 19. kinetics of ring-opening polymerization of ε-caprolactone initiated with functional aluminum alkoxides. Macromolecules 1996, 29, 1965–1975. [Google Scholar] [CrossRef]
- Zhang, G.Q.; Fiore, G.L.; Clair, T.S.; Fraser, C.L. Difluoroboron dibenzoylmethane PCL-PLA block copolymers: Matrix effects on room temperature phosphorescence. Macromolecules 2009, 42, 3162–3169. [Google Scholar] [CrossRef]
- Huang, W.Y.; Xue, X.Q.; Chen, J.H.; Fang, J.B.; Zhang, D.L.; Yang, H.J.; Jiang, B.B.; Kong, L.Z.; Pu, H.D. Preparation and characterisation of branched poly (styrene-co-acrylonitrile) via atom transfer radical polymerisation using β-bromoethyl benzene as initiator. Mater. Res. Innov. 2014, 18, 214–219. [Google Scholar] [CrossRef]
- Huang, W.Y.; Xue, X.Q.; Chen, J.H.; Fang, J.B.; Zhang, D.L.; Yang, H.J.; Jiang, B.B.; Kong, L.Z.; Liu, Y.; Pu, H.D. Influence of solvent on branching via atom transfer radical polymerisation using 1,6-bismaleimidohexane as branching agent. Mater. Res. Innov. 2014, 18, 220–224. [Google Scholar] [CrossRef]
- Yuan, Y.Y.; Wang, Y.C.; Du, J.Z.; Wang, J. Synthesis of amphiphilic ABC 3-miktoarm star terpolymer by combination of ring-opening polymerization and “click” chemistry. Macromolecules 2008, 41, 8620–8625. [Google Scholar] [CrossRef]
- Crescenzi, V.; Manzini, G.; Calzolari, G.; Borri, C. Thermodynamics of fusion of poly-β-propiolactone and poly-ε-caprolactone. comparative analysis of the melting of aliphatic polylactone and polyester chains. Eur. Polymer. J. 1972, 449–463. [Google Scholar] [CrossRef]
- Jia, Z.F.; Zhou, Y.F.; Yan, D.Y. Amphiphilic star-block copolymers based on a hyperbranched core: Synthesis and supramolecular self-assembly. J. Polym. Sci. Part A 2005, 43, 6543–6544. [Google Scholar] [CrossRef]
- Sin, S.L.; Gan, L.H.; Hu, X.; Tam, K.C.; Gan, Y.Y. Photochemical and thermal isomerizations of azobenzene-containing amphiphilic diblock copolymers in aqueous micellar aggregates and in film. Macromolecules 2005, 38, 3943–3948. [Google Scholar] [CrossRef]
- Zhao, Y. Design and study of new azobenzene liquidcrystal/polymer materials. Chin. J. Polym. Sci. 2003, 21, 621–629. [Google Scholar]
- Xu, Z.D.; Zhang, Y.; Fan, X.H.; Wan, X.H.; Zhou, Q.F. Optical phase conjugation response of photoinduced polymer films containing azobenzene moieties with chiral group. Chin. J. Polym. Sci. 2002, 20, 99–104. [Google Scholar]
- Erdogan, T.; Gungor, E.; Durmaz, H.; Hizal, G.; Tunca, U. Photoresponsive poly(methyl methacrylate)2-(polystyrene)2 miktoarm star copolymer containing an azobenzene moiety at the core. J. Polym. Sci. Part A 2006, 44, 1396–1403. [Google Scholar] [CrossRef]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Xue, X.; Yang, J.; Huang, W.; Yang, H.; Jiang, B. Synthesis of Hyperbranched Poly(ε-caprolactone) Containing Terminal Azobenzene Structure via Combined Ring-Opening Polymerization and “Click” Chemistry. Polymers 2015, 7, 1248-1268. https://doi.org/10.3390/polym7071248
Xue X, Yang J, Huang W, Yang H, Jiang B. Synthesis of Hyperbranched Poly(ε-caprolactone) Containing Terminal Azobenzene Structure via Combined Ring-Opening Polymerization and “Click” Chemistry. Polymers. 2015; 7(7):1248-1268. https://doi.org/10.3390/polym7071248
Chicago/Turabian StyleXue, Xiaoqiang, Jing Yang, Wenyan Huang, Hongjun Yang, and Bibiao Jiang. 2015. "Synthesis of Hyperbranched Poly(ε-caprolactone) Containing Terminal Azobenzene Structure via Combined Ring-Opening Polymerization and “Click” Chemistry" Polymers 7, no. 7: 1248-1268. https://doi.org/10.3390/polym7071248