
Perfluoro-p-xylene as a New Unique Monomer for Highly Stable Arylene Main-Chain Ionomers Applicable to Low-T and High-T Fuel Cell Membranes

Abstract
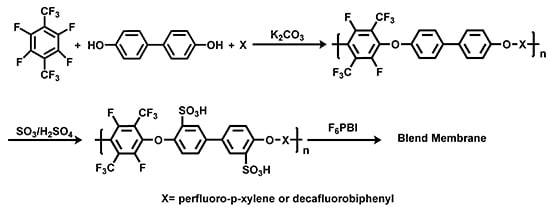
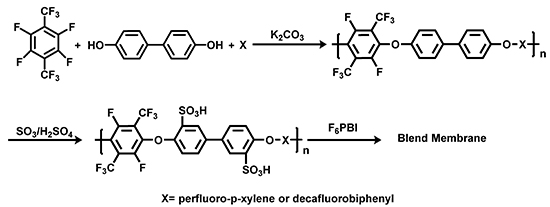
:
1. Introduction
2. Experimental Section
2.1. Materials
2.2. Synthesis
2.3. Blend Membrane Preparation
2.4. Instrumentation
3. Results and Discussion
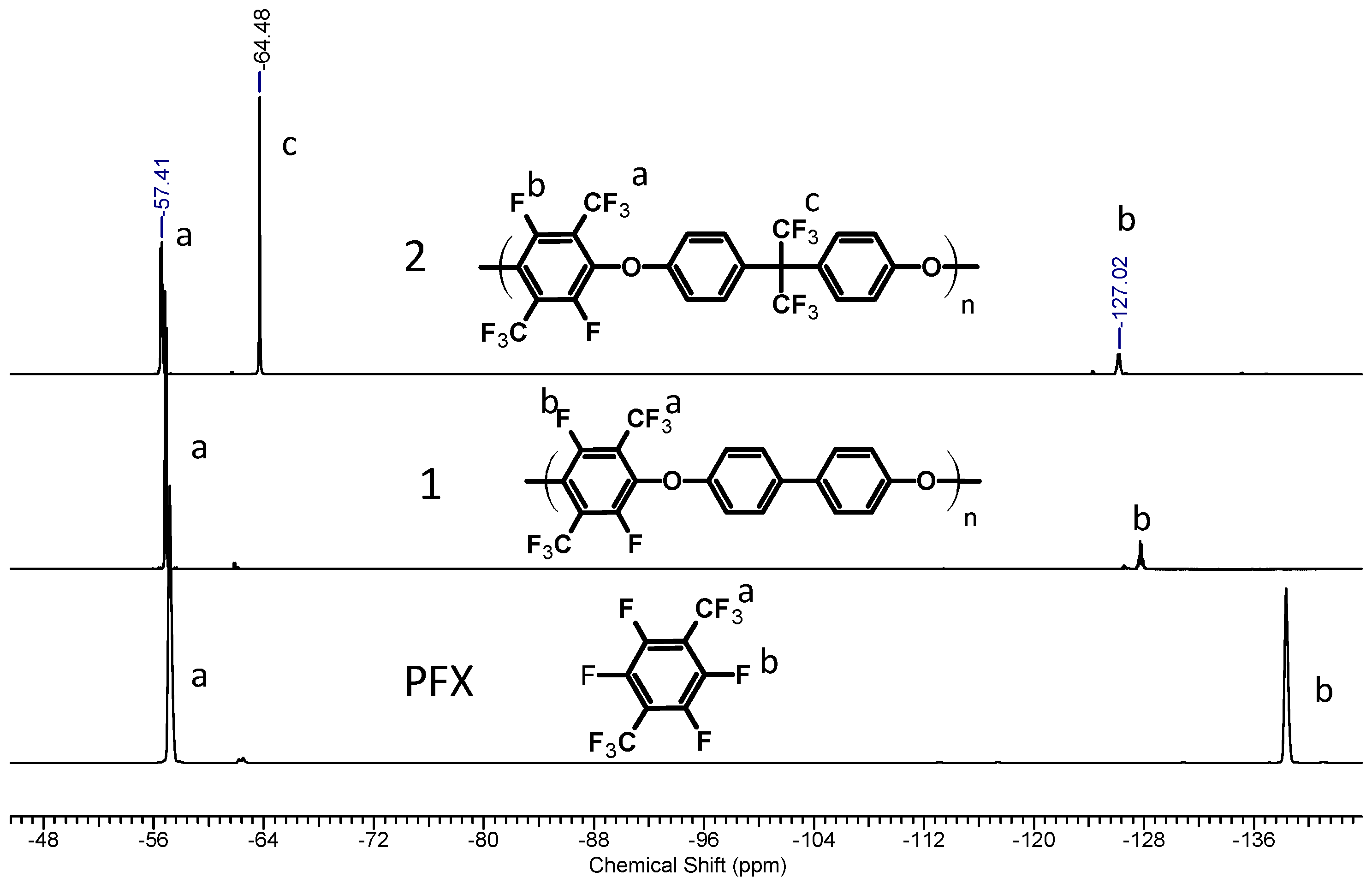
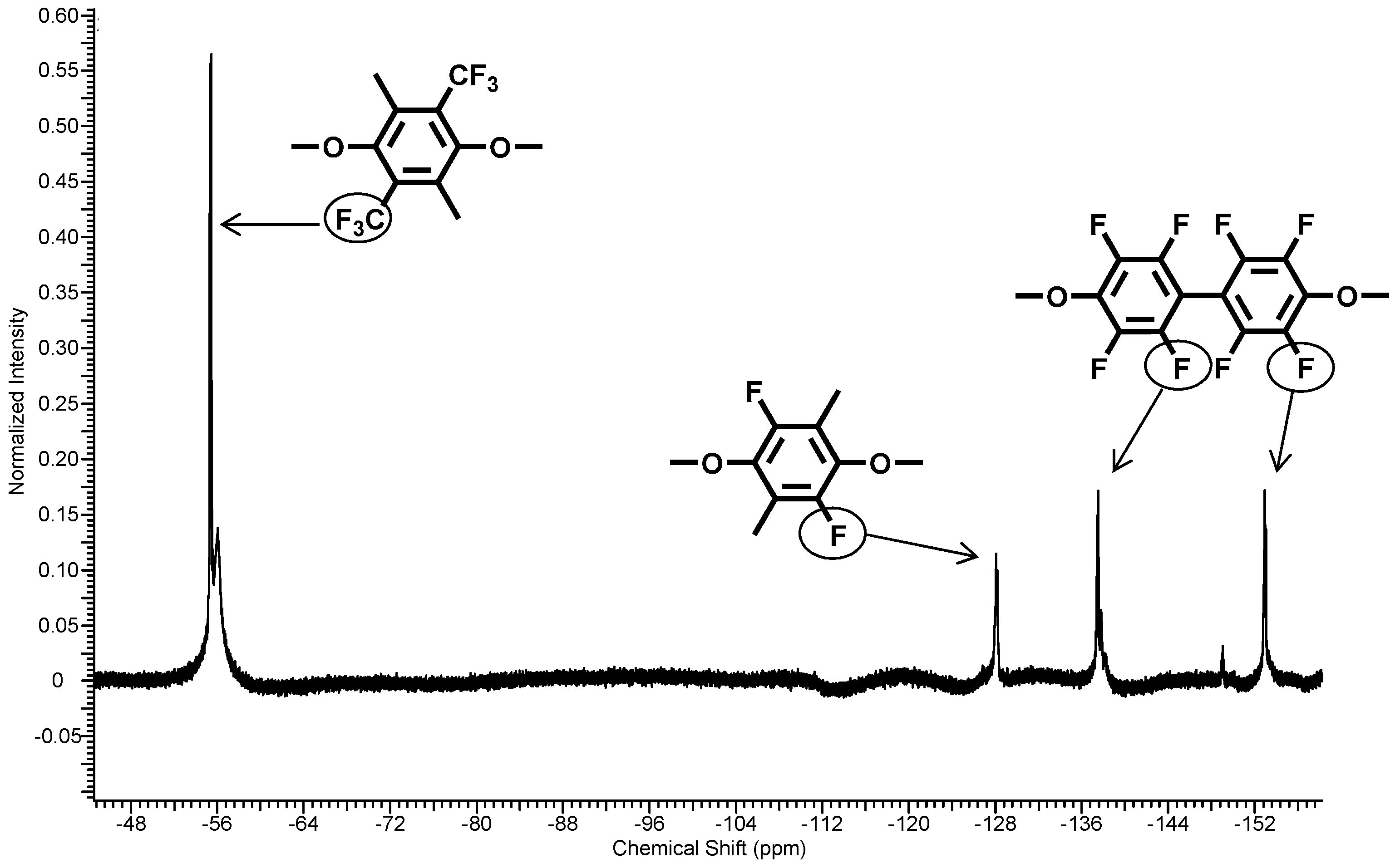
3.1. Polycondensation and Functionalization
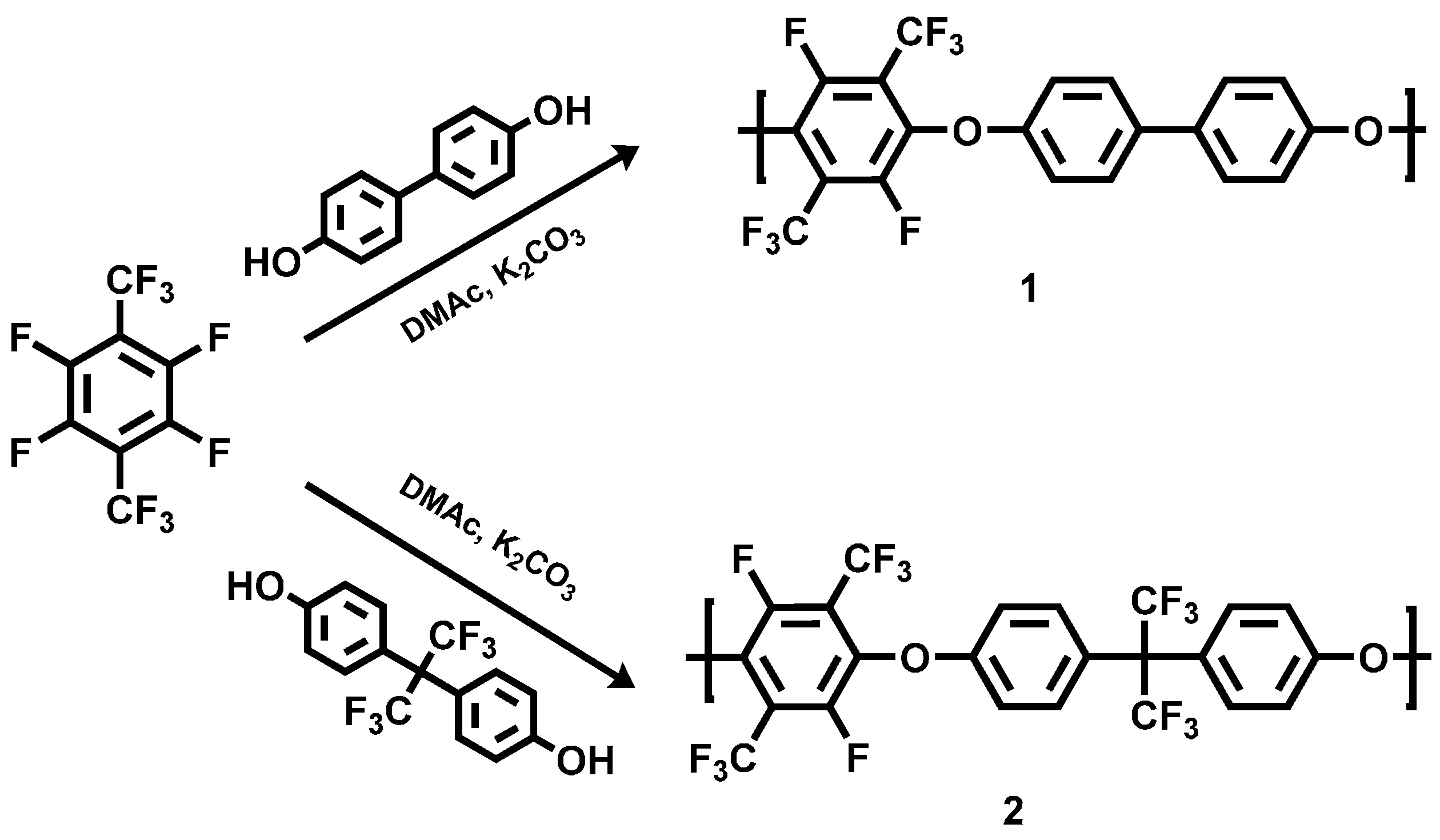
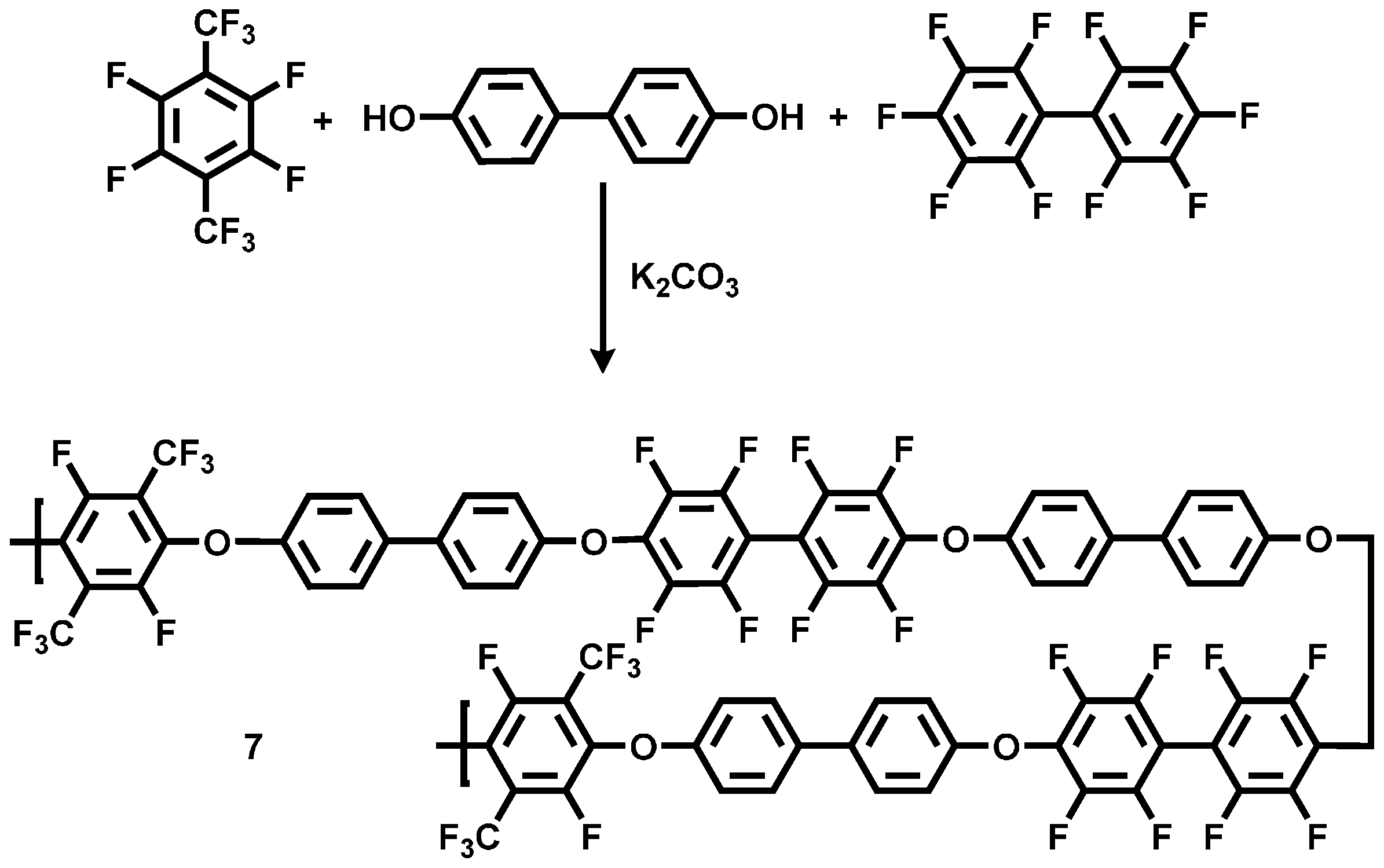
3.1.1. Polycondensation of Tetrafluoro-1,4-bis(trifluoromethyl)-benzene,4,4'-Dihydroxybiphenyl and 2,2-Bis(4-hydroxyphenyl)-hexafluoropropane



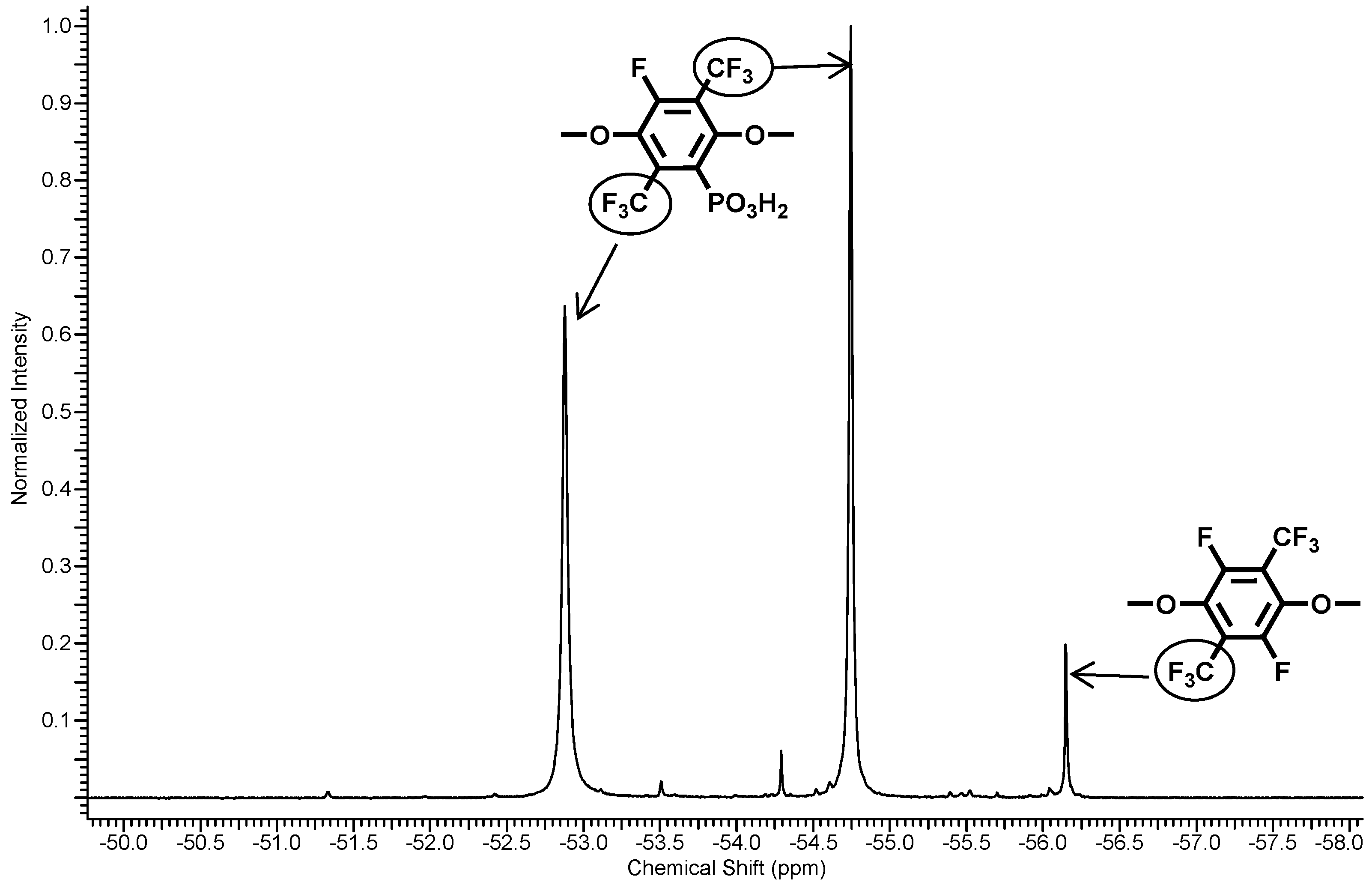
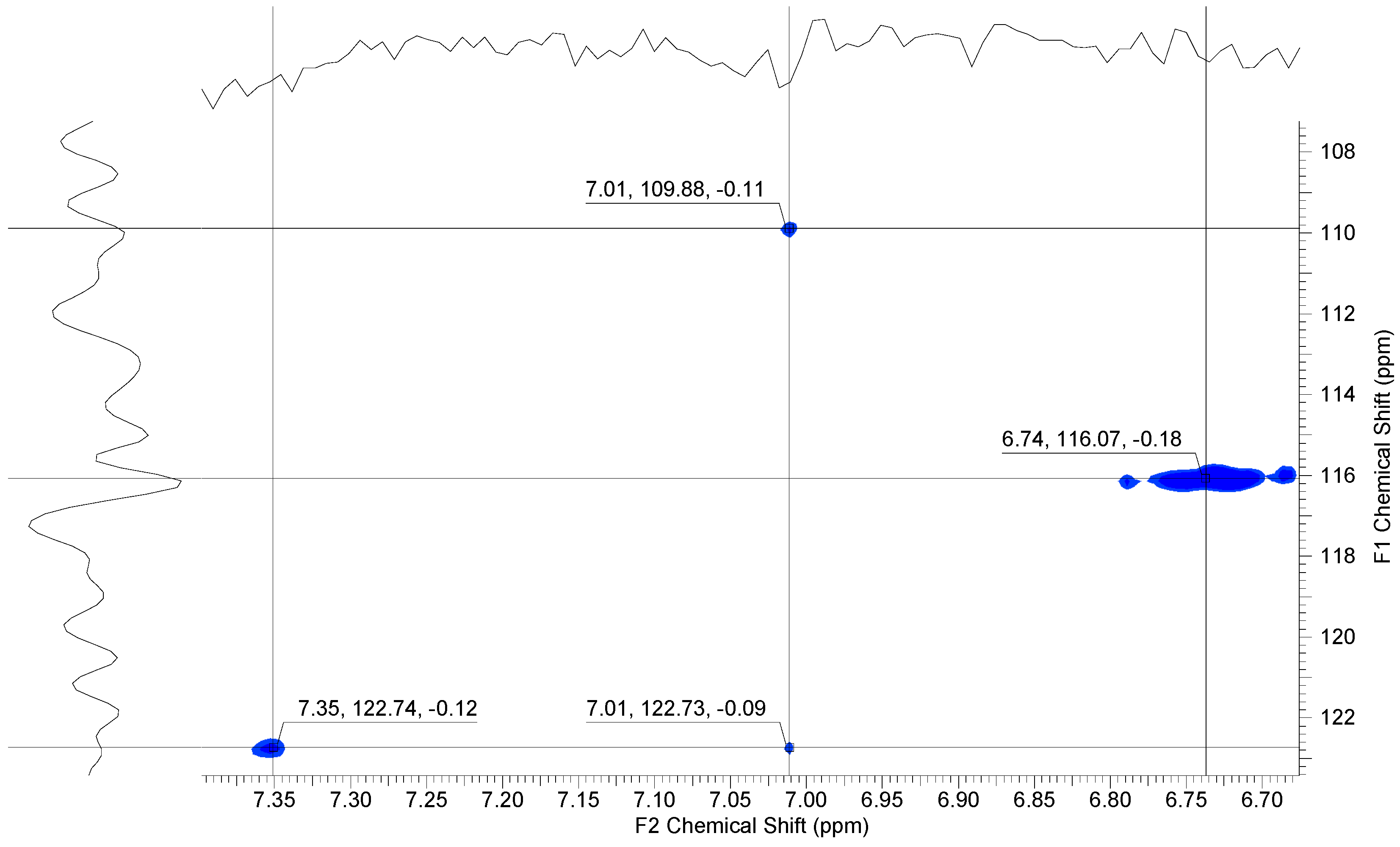
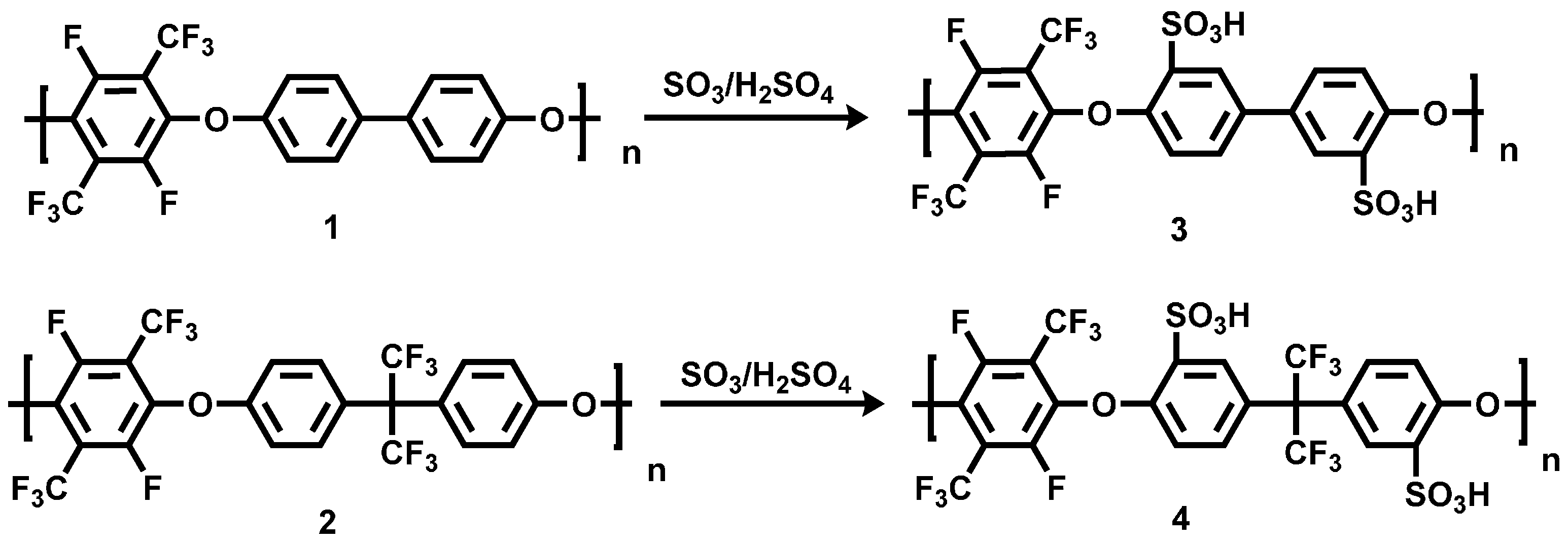
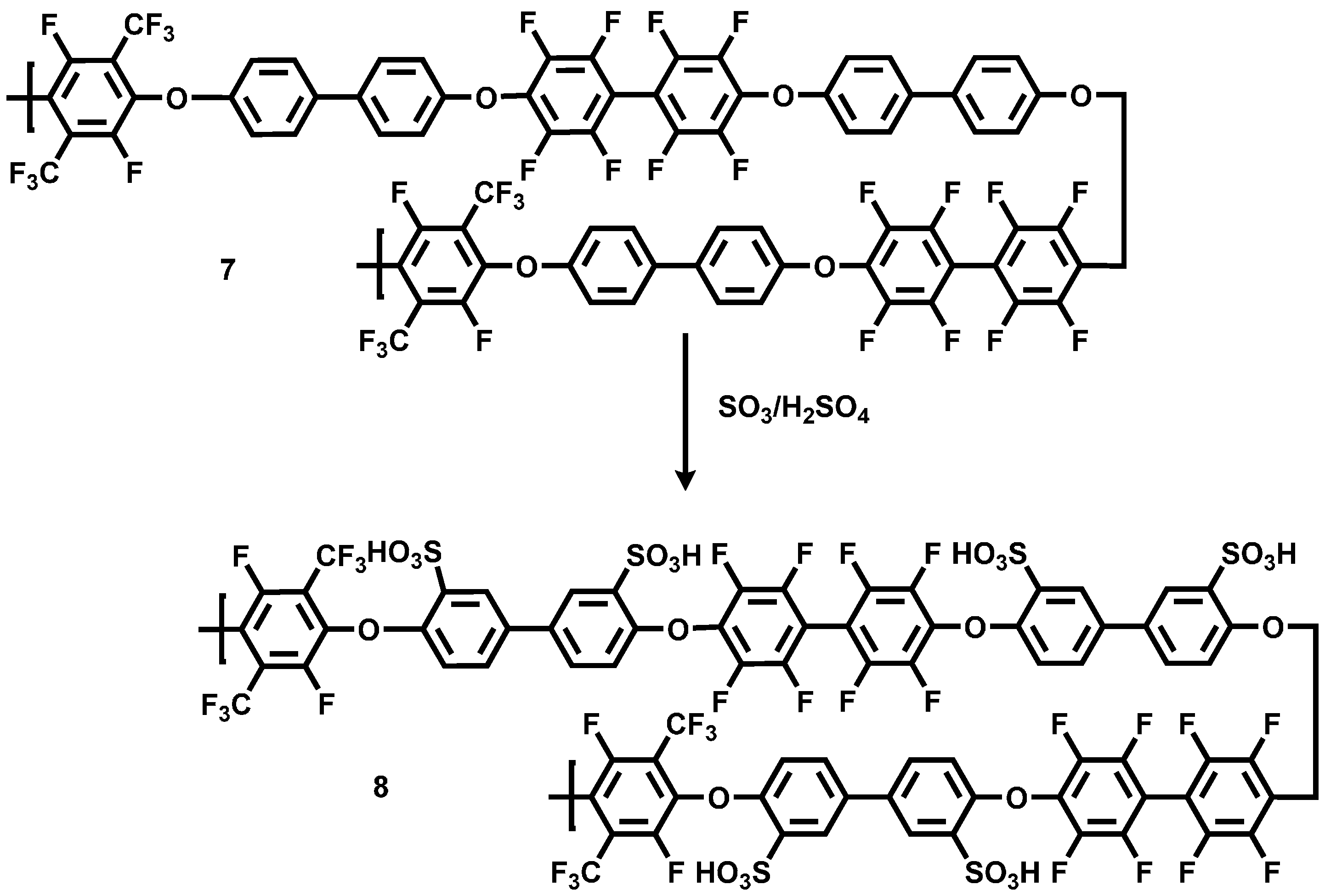
3.1.2. Sulfonation of Poly(arylene ether perfluoroxylene)s


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| pPFX (No.) | IECtheor (meq SO3H/g) | IECdirect (meq SO3H/g) | IECtotal (meq SO3H/g) |
|---|---|---|---|
| 3 | 3.30 | 2.43 | 2.80 |
| 4 | 2.68 | 2.01 | 2.60 |
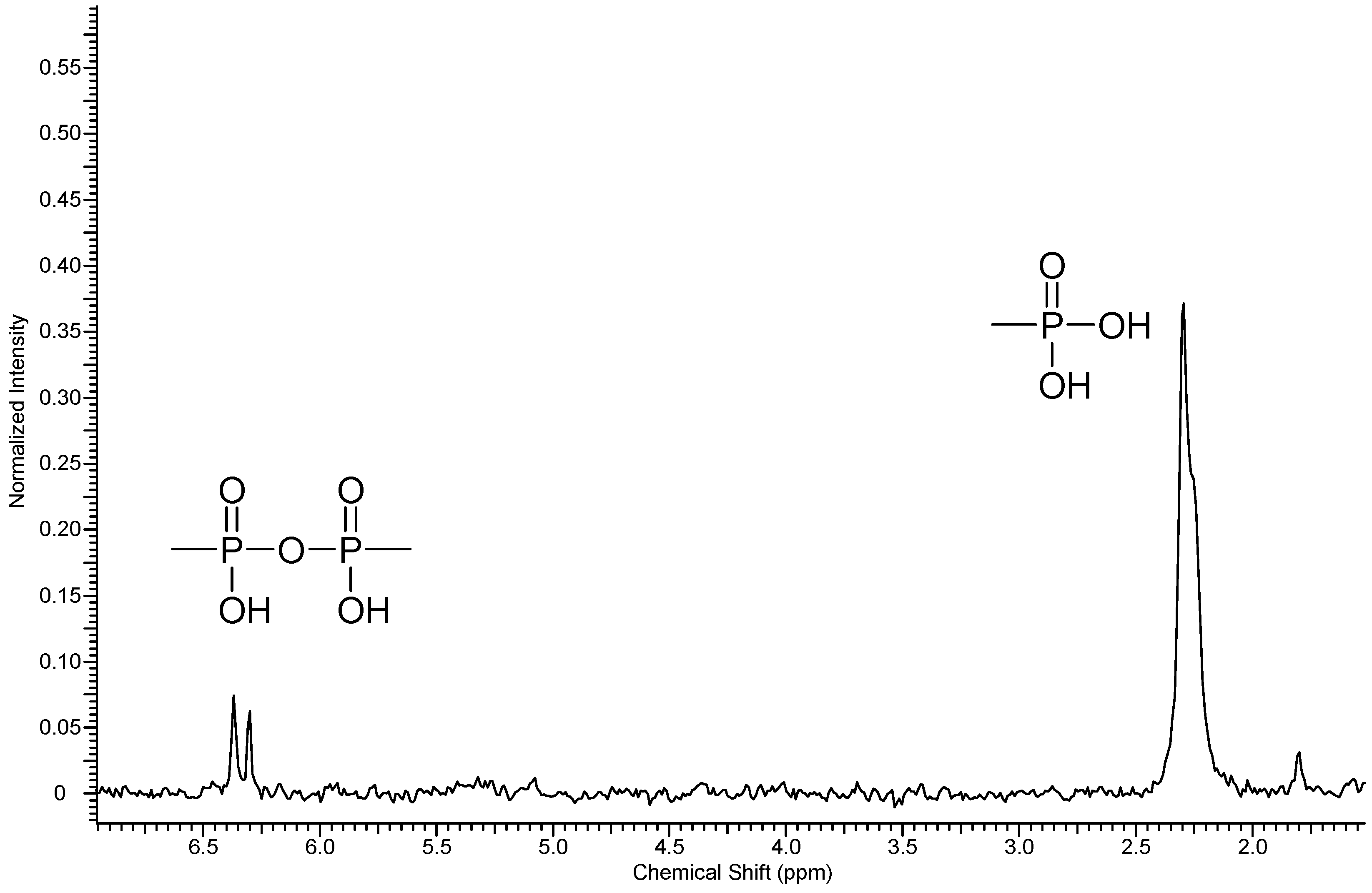
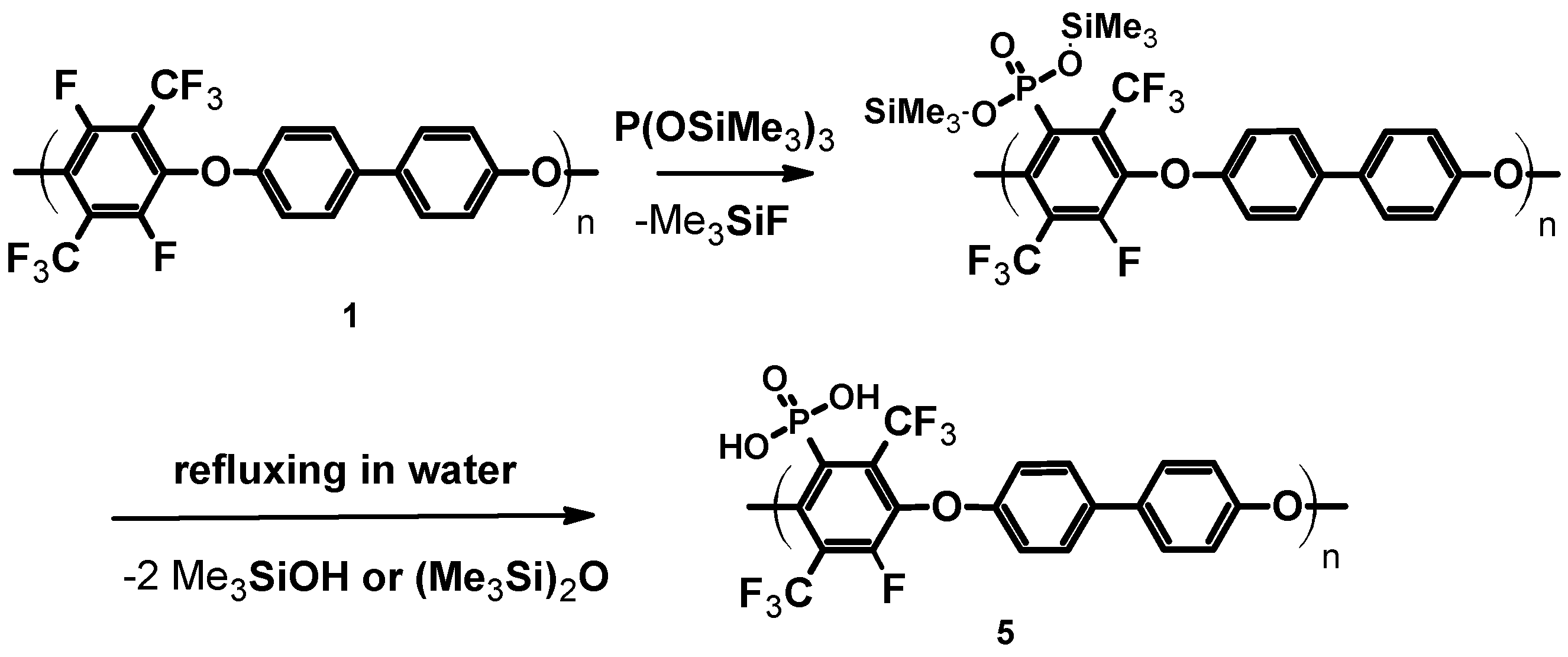
3.1.3. Phosphonation of Poly(arylene ether perfluoroxylene)



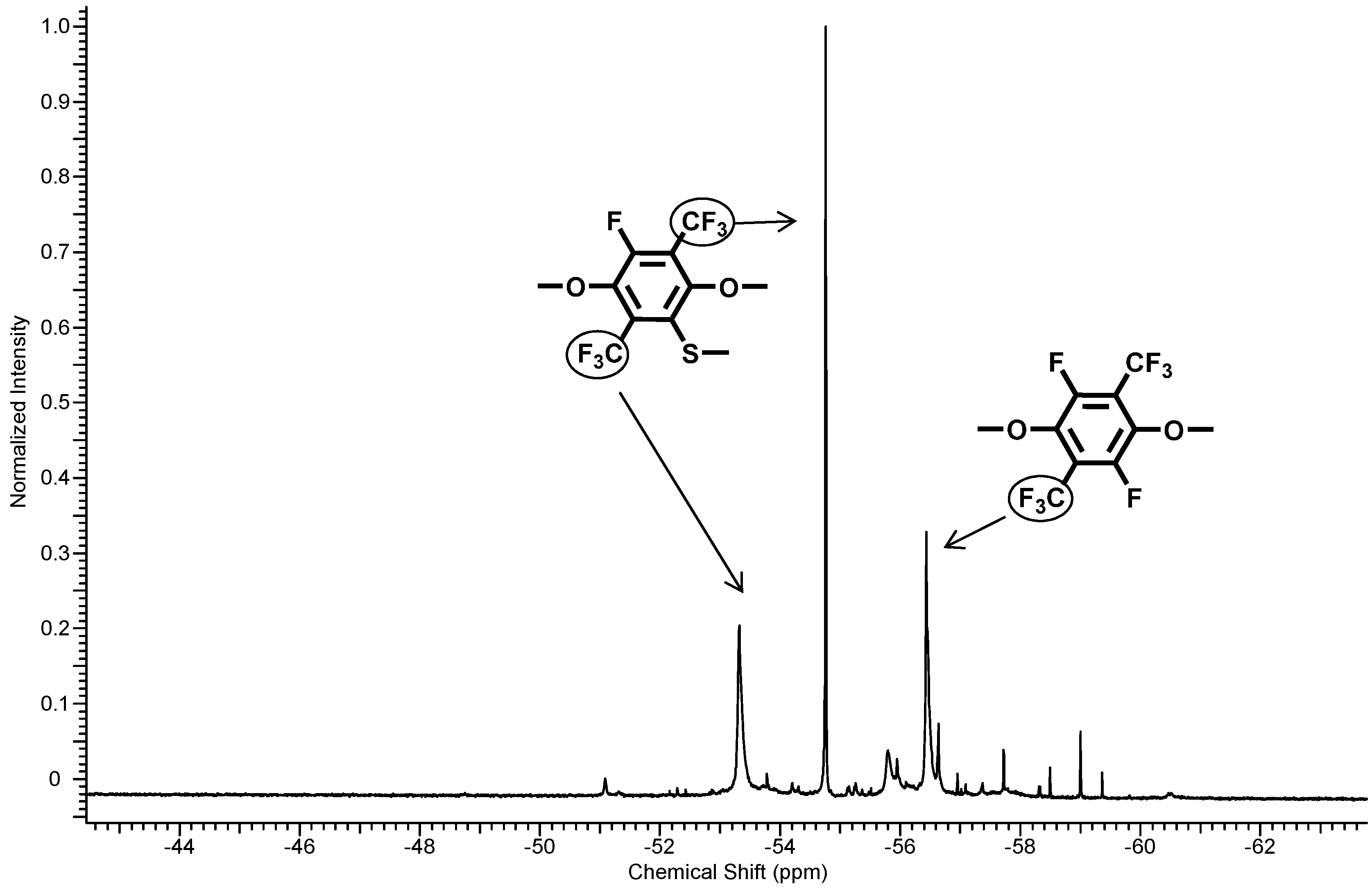
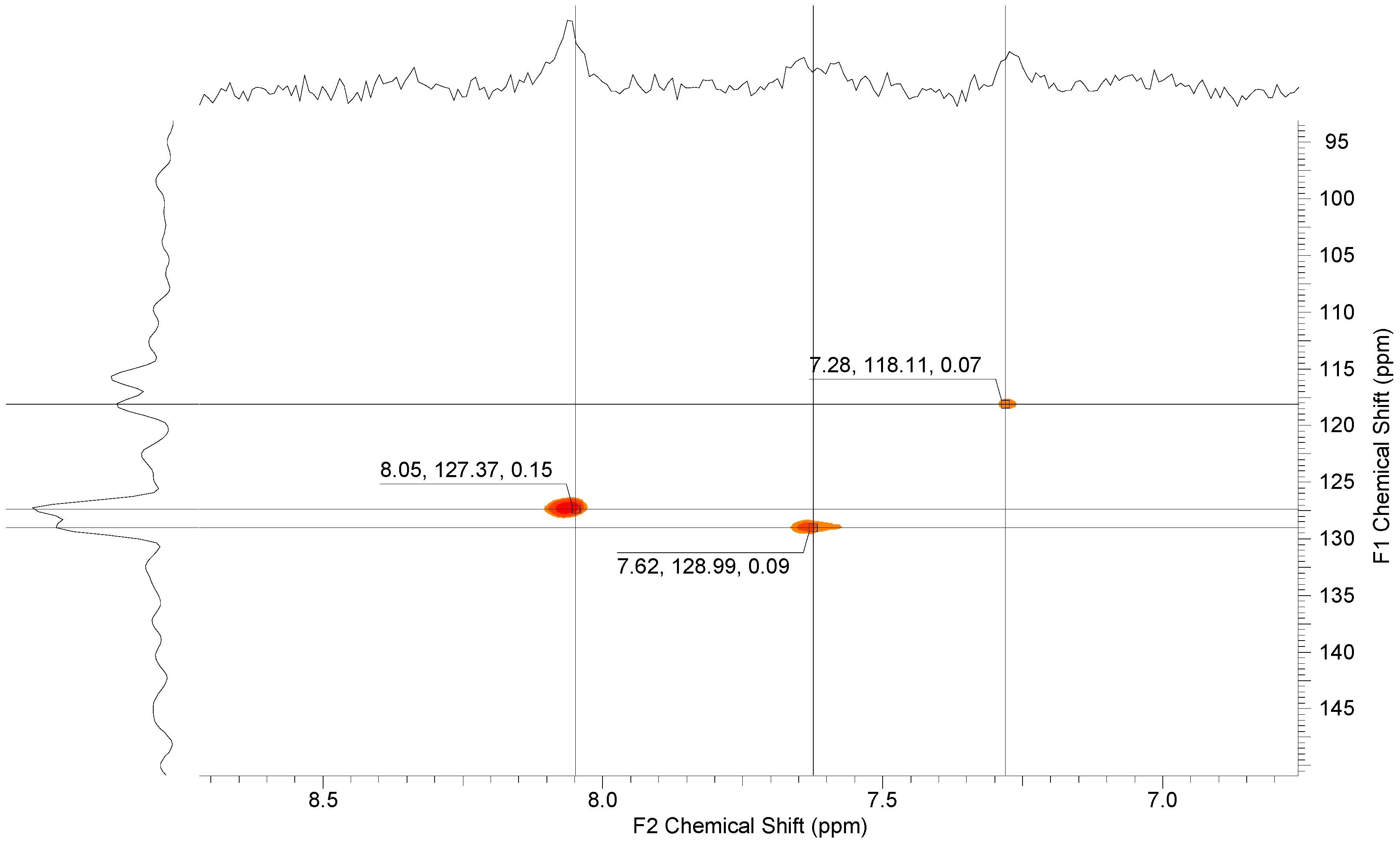
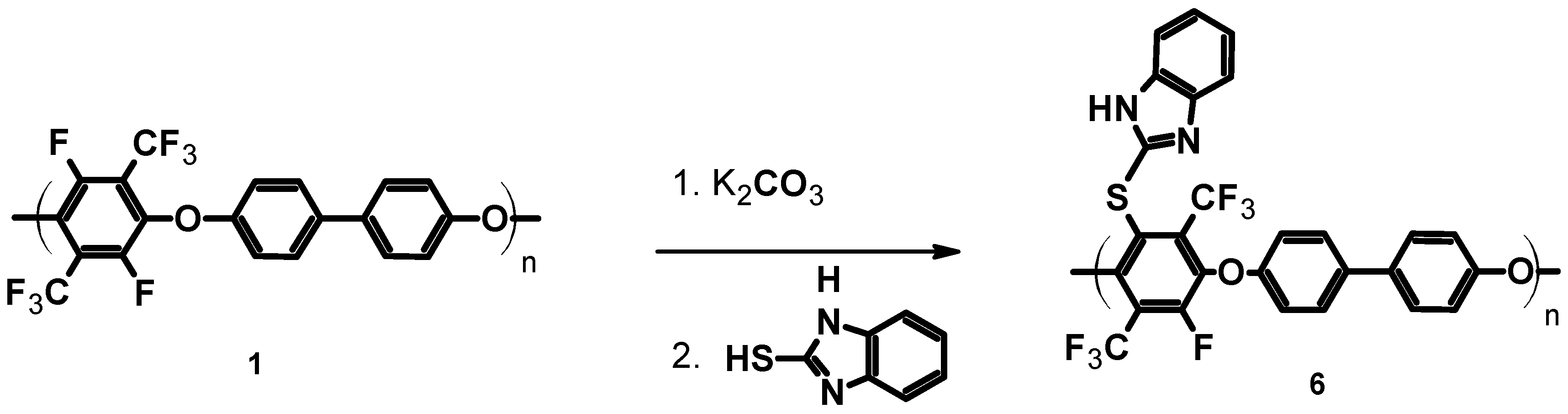
3.1.4. Functionalization of pPFX with Mercaptobenzimidazole



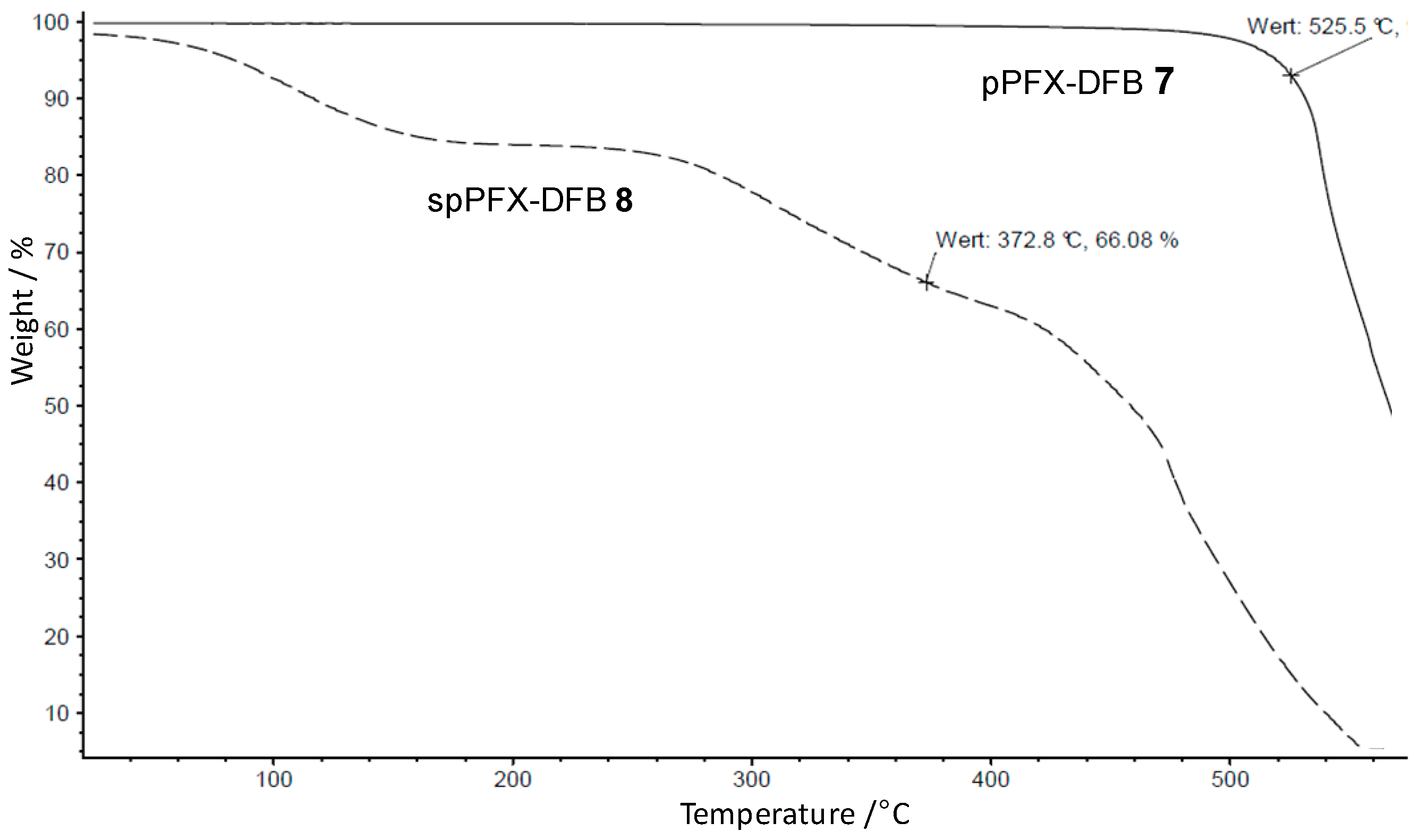
3.1.5. Terpolymer from PFX, Decafluorobiphenyl and 4,4'-Dihydroxy-biphenyl



3.1.6. Sulfonation of pPFX-DFB with Fuming Sulfuric Acid


| pPFX (No.) | IECtheor (meq SO3H/g) | IECdirect (meq SO3H/g) | IECtotal (meq SO3H/g) |
|---|---|---|---|
| 8 | 1.87 | 1.39 | 1.72 |
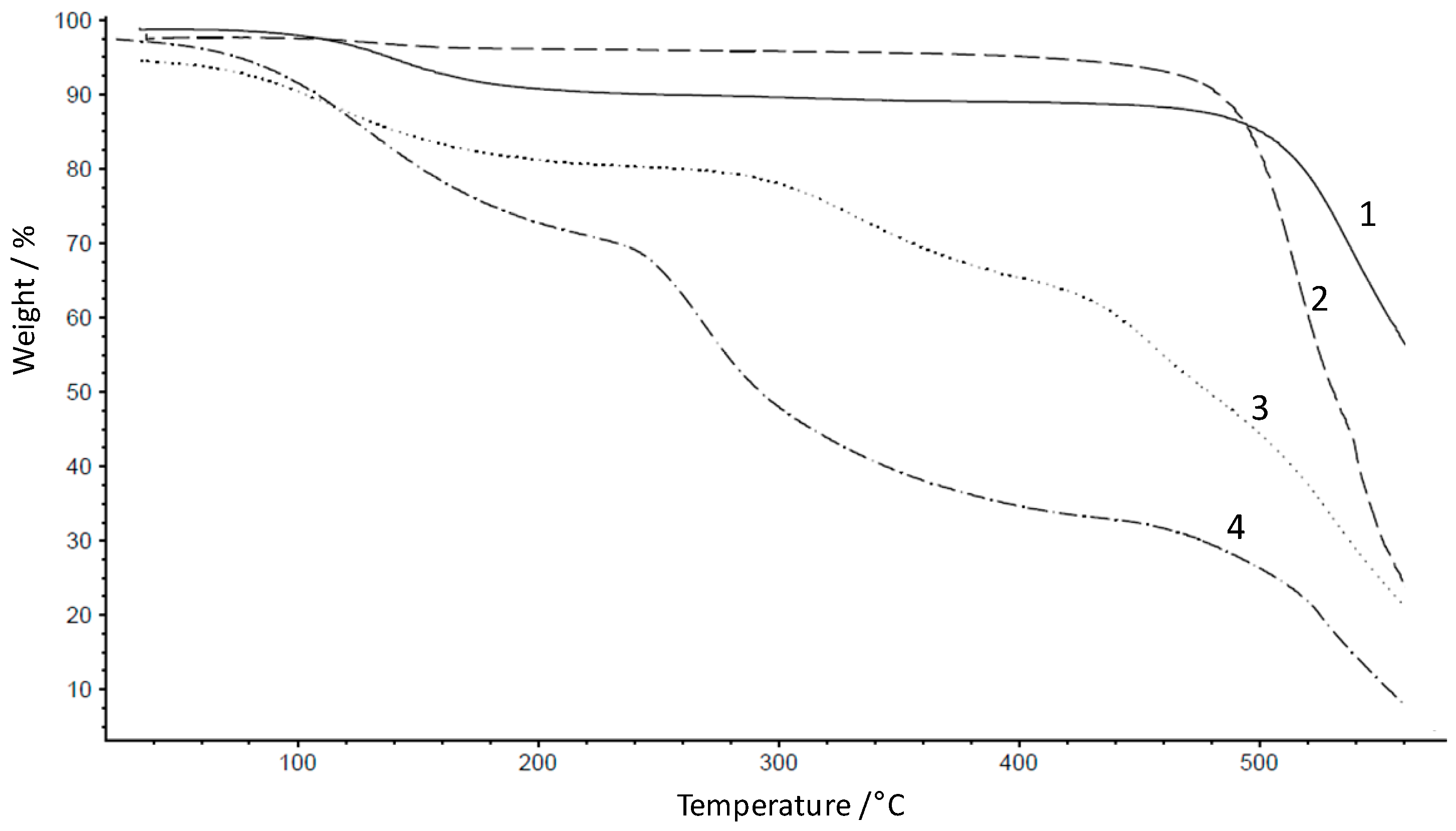
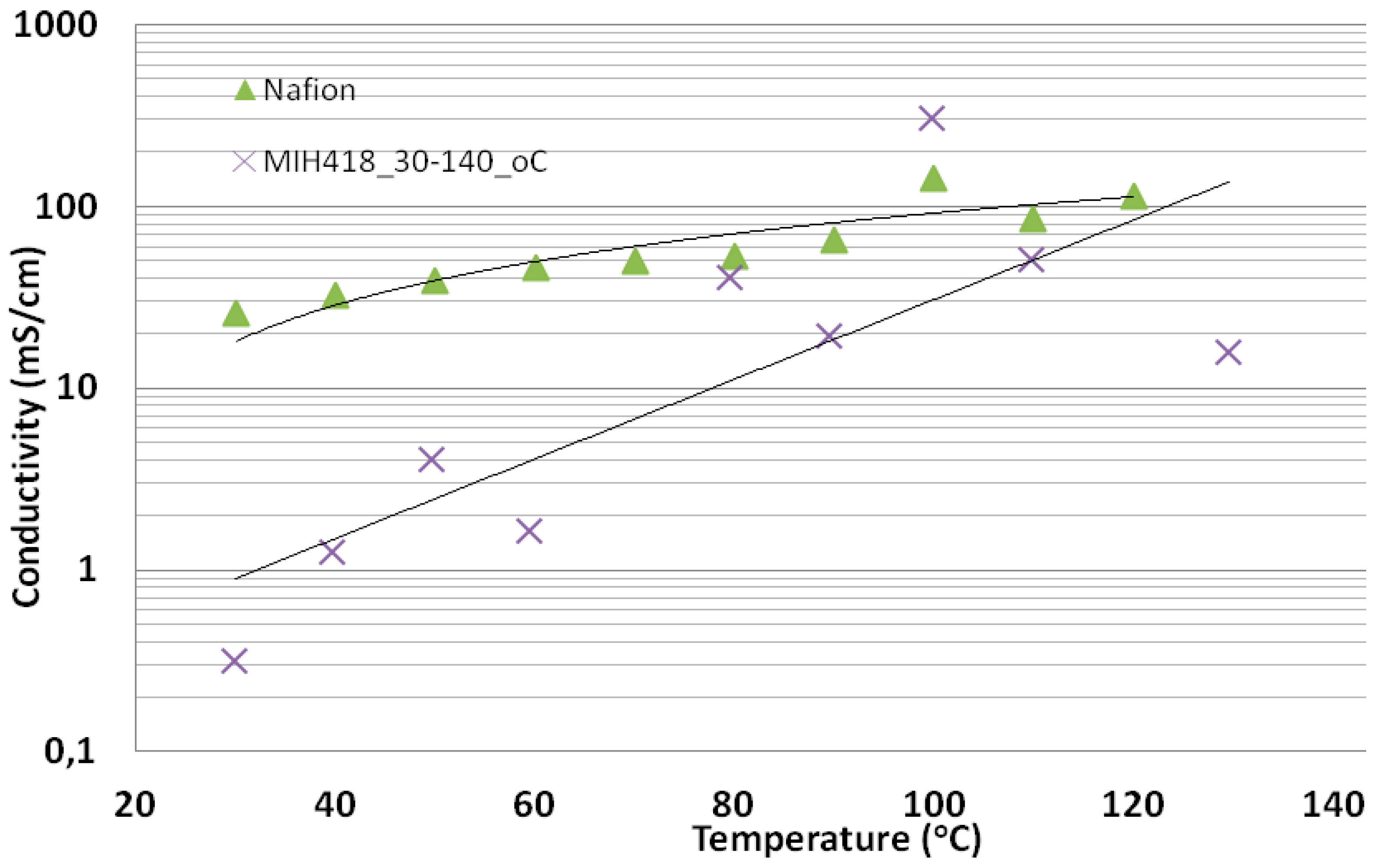
3.2. Preparation and Properties of Acid-Excess Acid-Base Blend Membranes MIH418
| Nr. | spPFX-DFB (wt%) | F6PBI (wt%) | Thickn. (nm) | IECdirect | IECtotal | Tdecomp. (°C) * |
|---|---|---|---|---|---|---|
| MIH 418 | 80 | 20 | 160 | 1.33 | 1.83 | 424.5 |
Conductivity of the Acid Excess Acid-Base Blend Membrane MIH418

3.3. Preparation and Properties of Base-Excess Acid-Base Blend Membranes
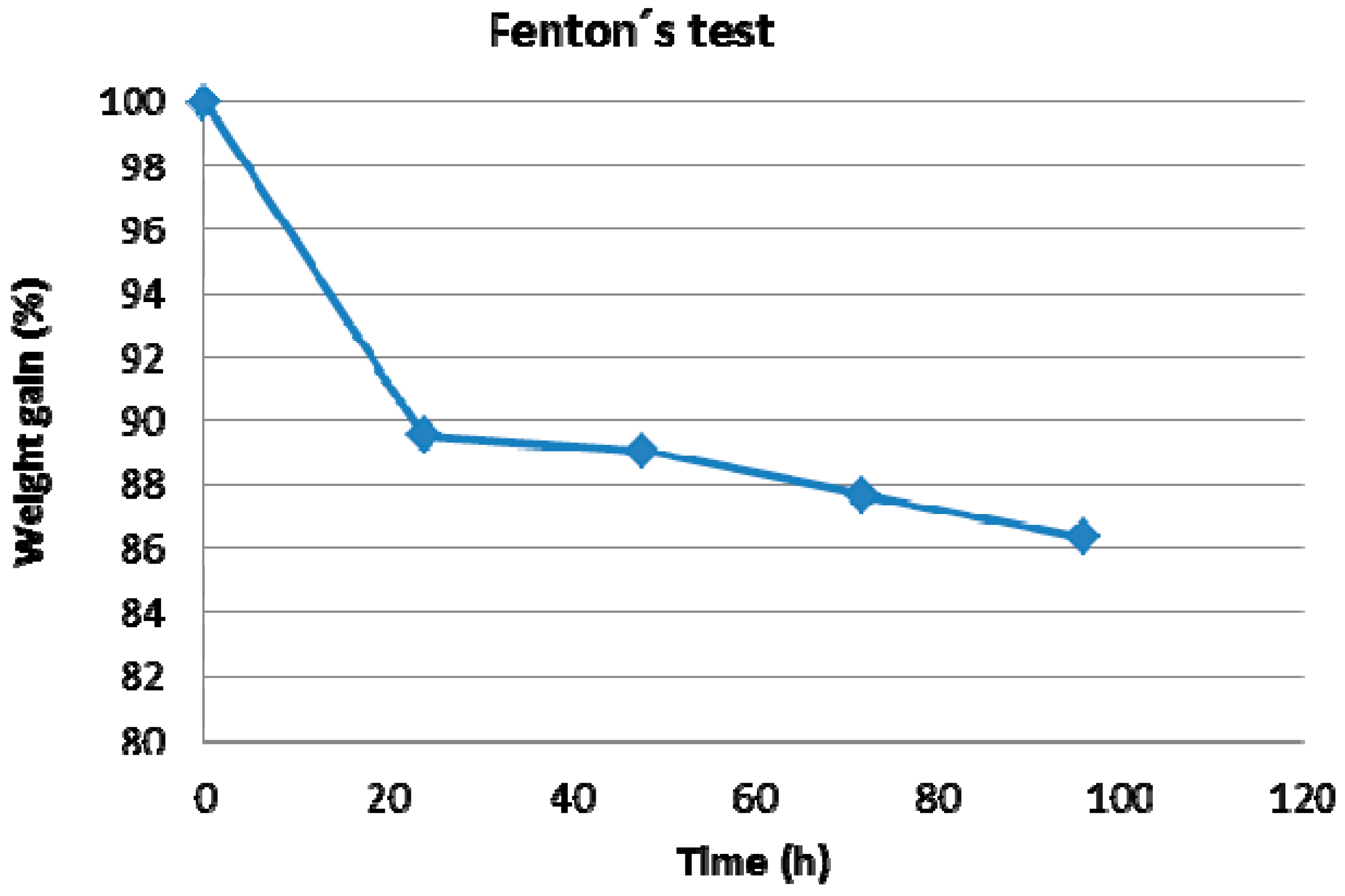
3.3.1. Oxidative Stability of the Base-Excess Acid-Base Blend Membranes

3.3.2. Phosphoric Acid Doping of the Base-Excess Blend Membrane
3.3.3. Conductivity of the H3PO4-Doped F6-PBI Blend Membranes

4. Conclusions
Supplementary materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Liu, B.; Robertson, G.P.; Kim, D.S.; Guiver, M.D.; Hu, W.; Jiang, Z. Aromatic poly(ether ketone)s with pendant sulfonic acid phenyl groups prepared by a mild sulfonation method for proton exchange membranes. Macromolecules 2007, 40, 1934–1944. [Google Scholar] [CrossRef]
- Gautier-Luneau, I.; Denoyelle, A.; Sanchez, J.Y.; Poinsignon, C. Organic-inorganic protonic polymer electrolytes as membrane for low-temperature fuel cell. Electrochim. Acta 1992, 37, 1615–1618. [Google Scholar] [CrossRef]
- Allcock, H.R.; Hofmann, H.A.; Ambler, C.M.; Morford, R.V. Phenylphosphonic acid functionalized Poly[aryloxyphosphazenes]. Macromolecules 2002, 35, 3484–3489. [Google Scholar] [CrossRef]
- Allcock, H.R.; Wood, R.M. Design and synthesis of ion-conductive polyphosphazenes for fuel cell applications: Review. J. Polym. Sci. B Polym. Phys. 2006, 44, 2358–2368. [Google Scholar] [CrossRef]
- Gubler, R.; Beck, N.; Gürsel, S.A.; Hajbolouri, F.; Kramer, D.; Reiner, A.; Steiger, B.; Scherer, G.; Wokaun, A.; Rajesh, B.; et al. Materials for polymer electrolyte fuel cells. Chimia 2004, 58, 826–836. [Google Scholar] [CrossRef]
- Schönberger, F.; Hein, M.; Kerres, J. Preparation and characterisation of sulfonated partially fluorinated statistical poly(arylene ether sulfone)s and their blends with PBI. Solid State Ion. 2007, 178, 547–554. [Google Scholar] [CrossRef]
- Kerres, J.A.; Xing, D.; Schönberger, F. Comparative investigation of novel PBI blend ionomer membranes from nonfluorinated and partially fluorinated poly arylene ethers. J. Polym. Sci. B Polym. Phys. 2006, 44, 2311–2326. [Google Scholar] [CrossRef]
- Kerres, J.; Ullrich, A.; Meier, F.; Häring, T. Synthesis and characterization of novel acid–base polymer blends for application in membrane fuel cells. Solid State Ion. 1999, 125, 243–249. [Google Scholar] [CrossRef]
- Kerres, J.; Ullrich, A.; Häring, T.; Baldauf, M.; Gebhardt, U.; Preidel, W. Preparation, characterization and fuel cell application of new acid-base blend membranes. J. New Mater Electrochem. Syst. 2000, 3, 229–239. [Google Scholar]
- Cooper, K.R. Progress toward accurate through-plane ion transport resistance measurement of thin solid electrolytes. J. Electrochem. Soc. 2010, 157, B1731–B1739. [Google Scholar] [CrossRef]
- Ding, J.; Day, M. Novel highly fluorinated poly(arylene ether-1,3,4-oxadiazole)s, their preparation, and sensory properties to fluoride anion. Macromolecules 2006, 39, 6054–6062. [Google Scholar] [CrossRef]
- Hajdok, I.; Bona, A.; Werner, H.; Kerres, J. Synthesis and characterization of fluorinated and sulfonated poly(arylene ether-1,3,4-Oxadiazole) derivatives and their blend membranes. Eur. Polym. J. 2014, 52, 76–87. [Google Scholar] [CrossRef]
- Kerres, J.; Cui, W.; Reichle, S. New sulfonated engineering polymers via the metalation route. I. Sulfonated poly(ethersulfone) PSU Udel® via metalation-sulfination-oxidation. J. Polym. Sci. A Polym. Chem. 1996, 34, 2421–2438. [Google Scholar] [CrossRef]
- Beletskaya, I.P.; Artamkina, G.A.; Milchenko, A.Y.; Sazonov, P.K.; Shtern, M.M. Carbonylmetallates and carbanions in aromatic and vinylic nucleophilic substitution. J. Phys. Org. Chem. 1996, 9, 319–328. [Google Scholar] [CrossRef]
- Miller, A.O.; Furin, G.G. N-(polyfluoroaryl)-hydroxylamines. synthesis and properties. J. Fluor. Chem. 1987, 36, 247–272. [Google Scholar] [CrossRef]
- Wang, Y.; Parkin, S.R.; Watson, M.D. Benzodichalcogenophenes with perfluoroarene termini. Org. Lett. 2008, 10, 4420–4424. [Google Scholar] [CrossRef] [PubMed]
- Atanasov, V.; Kerres, J. Highly phosphonated polypentafluorostyrene. Macromolecules 2011, 44, 6416–6423. [Google Scholar] [CrossRef]
- Kaltbeitzel, A.; Schauff, S.; Steininger, H.; Bingol, B.; Brunklaus, G.; Meyer, W.H.; Spiess, H.W. Water sorption of poly(vinylphosphonic acid) and its influence on proton conductivity. Solid State Ion. 2007, 178, 469–474. [Google Scholar] [CrossRef]
- Ohms, G.; Großmann, G.; Schwab, B.; Schiefer, H. Synthesis and 31P and 13C NMR studies of pyrophosphonic acid. Phosphorus Sulfur Silicon 1992, 68, 77–89. [Google Scholar] [CrossRef]
- Piergies, N.; Proniewicz, E. Structure characterization of [N-Phenylamino(2-boronphenyl)-R-methyl]phosphonic acid by vibrational spectroscopy and density functional theory calculations. J. Spectrosc. 2014, 8, 1–8. [Google Scholar] [CrossRef]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hajdok, I.; Atanasov, V.; Kerres, J. Perfluoro-p-xylene as a New Unique Monomer for Highly Stable Arylene Main-Chain Ionomers Applicable to Low-T and High-T Fuel Cell Membranes. Polymers 2015, 7, 1066-1087. https://doi.org/10.3390/polym7061066
Hajdok I, Atanasov V, Kerres J. Perfluoro-p-xylene as a New Unique Monomer for Highly Stable Arylene Main-Chain Ionomers Applicable to Low-T and High-T Fuel Cell Membranes. Polymers. 2015; 7(6):1066-1087. https://doi.org/10.3390/polym7061066
Chicago/Turabian StyleHajdok, Imre, Vladimir Atanasov, and Jochen Kerres. 2015. "Perfluoro-p-xylene as a New Unique Monomer for Highly Stable Arylene Main-Chain Ionomers Applicable to Low-T and High-T Fuel Cell Membranes" Polymers 7, no. 6: 1066-1087. https://doi.org/10.3390/polym7061066