Electrochemical and Spectroelectrochemical Properties of a New Donor–Acceptor Polymer Containing 3,4-Dialkoxythiophene and 2,1,3-Benzothiadiazole Units

Abstract
:1. Introduction

2. Experimental Section
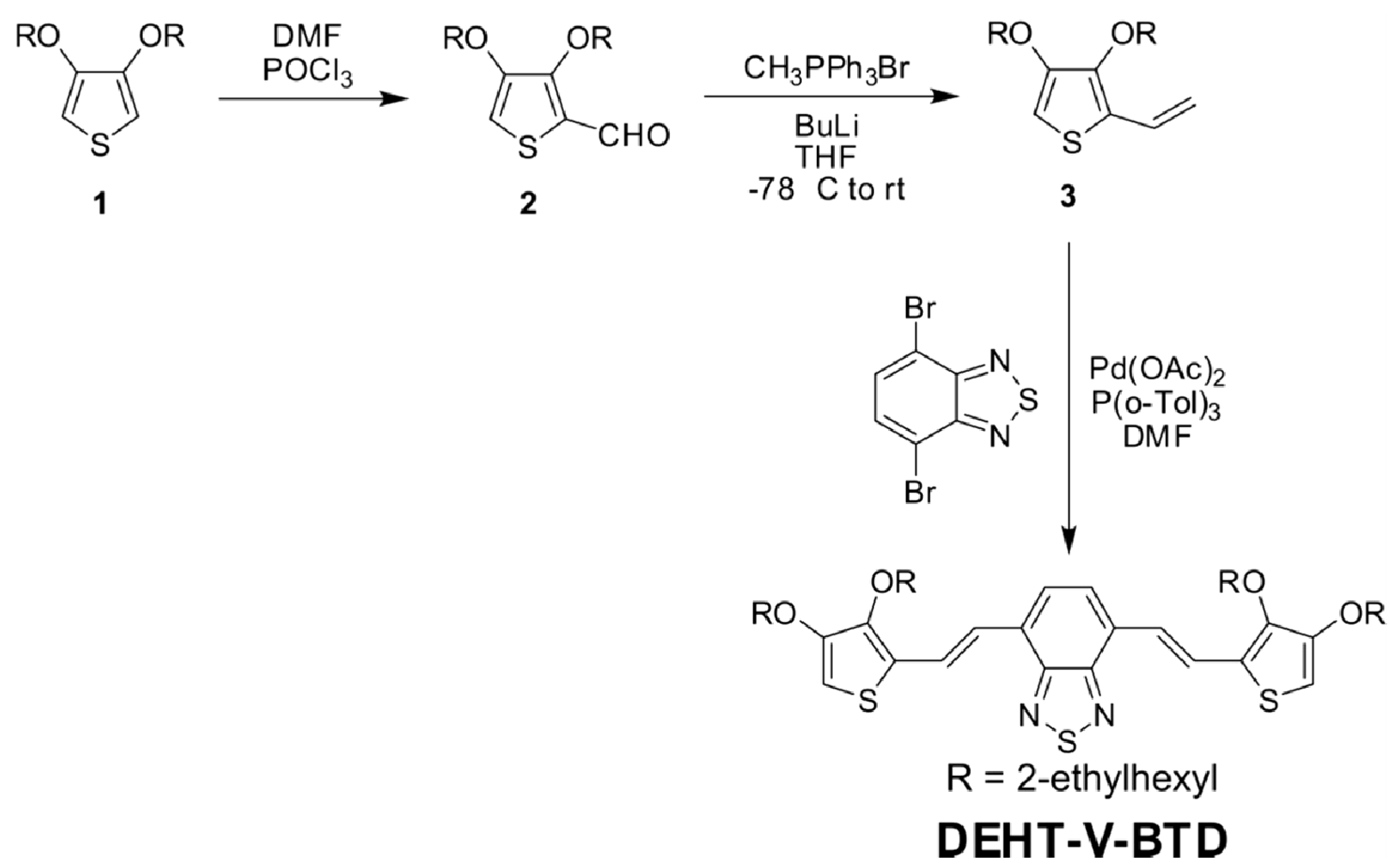
2.1. Monomer Preparation
2.2. Optical Characterization
2.3. Electropolymerization and Electrochemical Characterizations
3. Results and Discussion
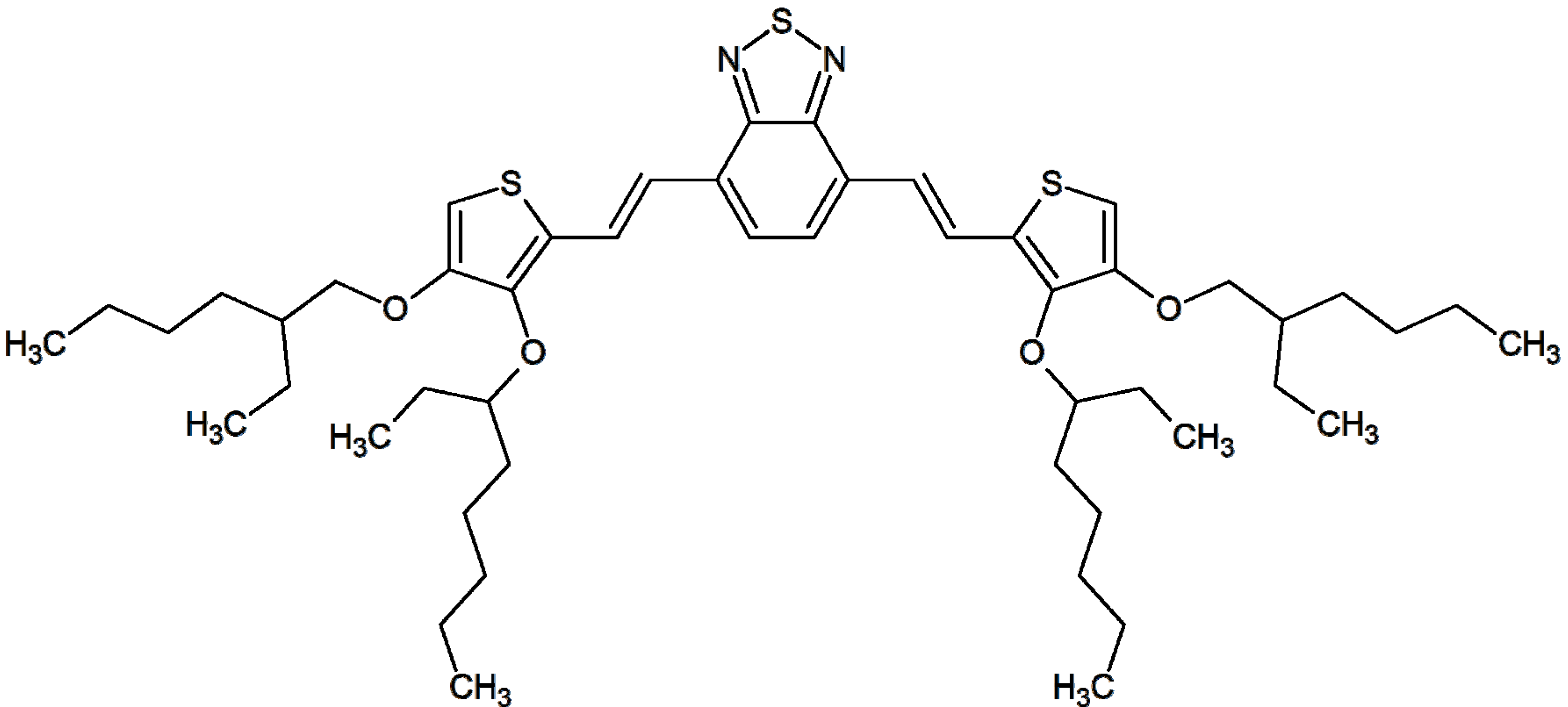
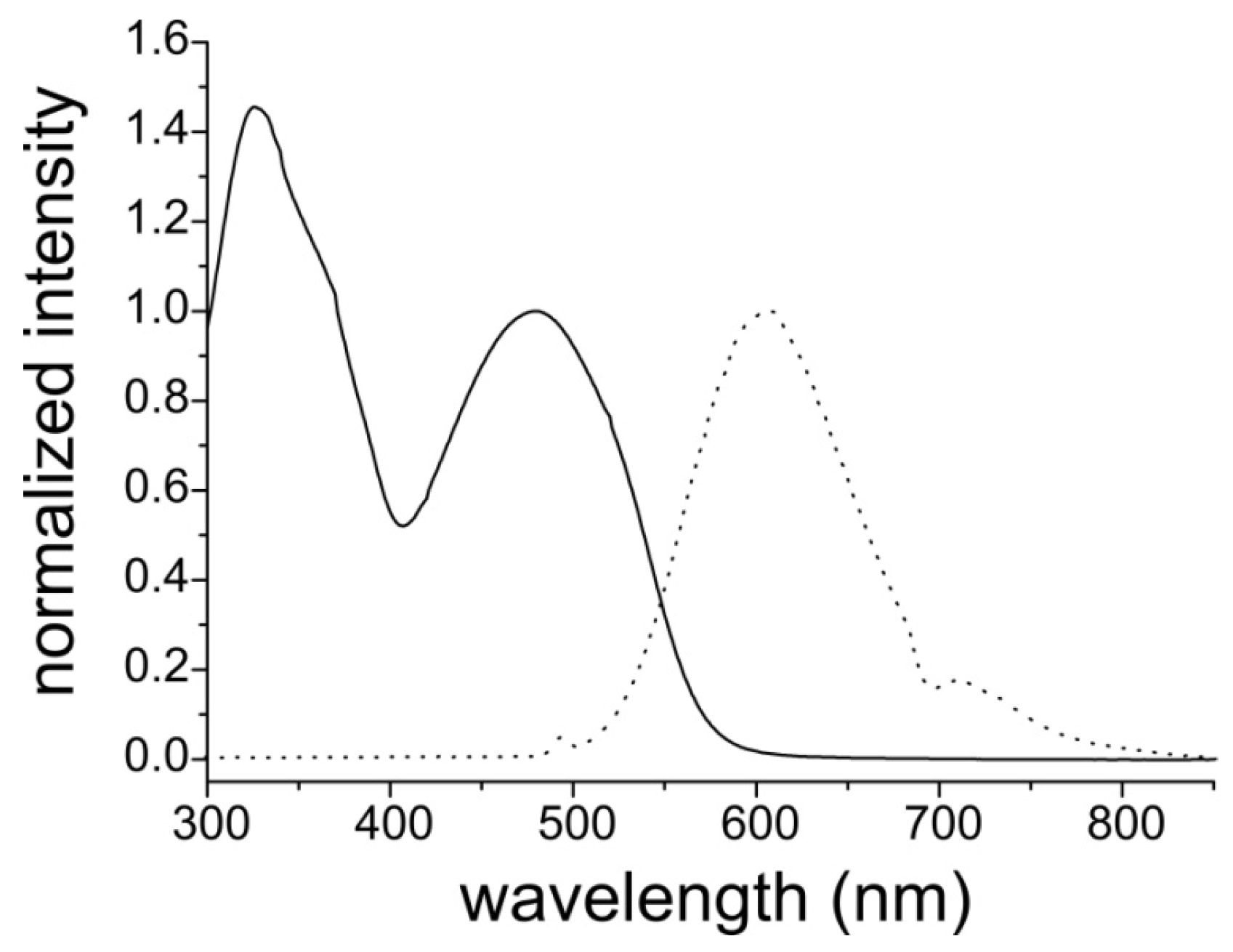
3.1. Synthesis and Optical Properties of the Monomer



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Species | Egapopt a [eV] | Eonsetox b [V] | HOMO c [eV] | Eonsetred b [V] | LUMO c [eV] | Egapel d [eV] |
|---|---|---|---|---|---|---|
| DEHT-V-BTD | 2.2 | 0.33 | −5.5 | – | – | – |
| Poly(DEHT-V-BTD) | 1.8 | 0.15 | −5.3 | −1.60 | −3.6 | 1.7 |
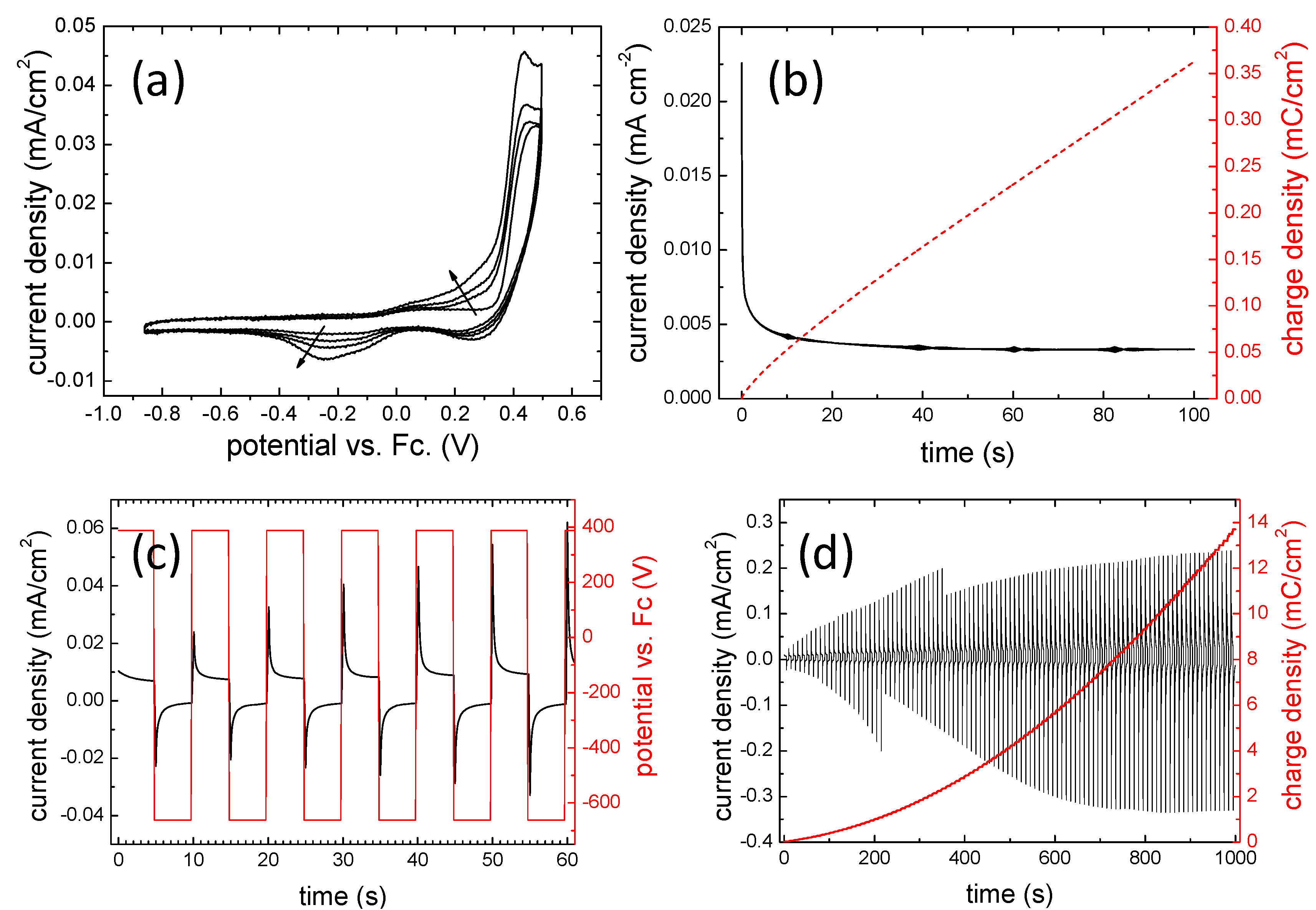
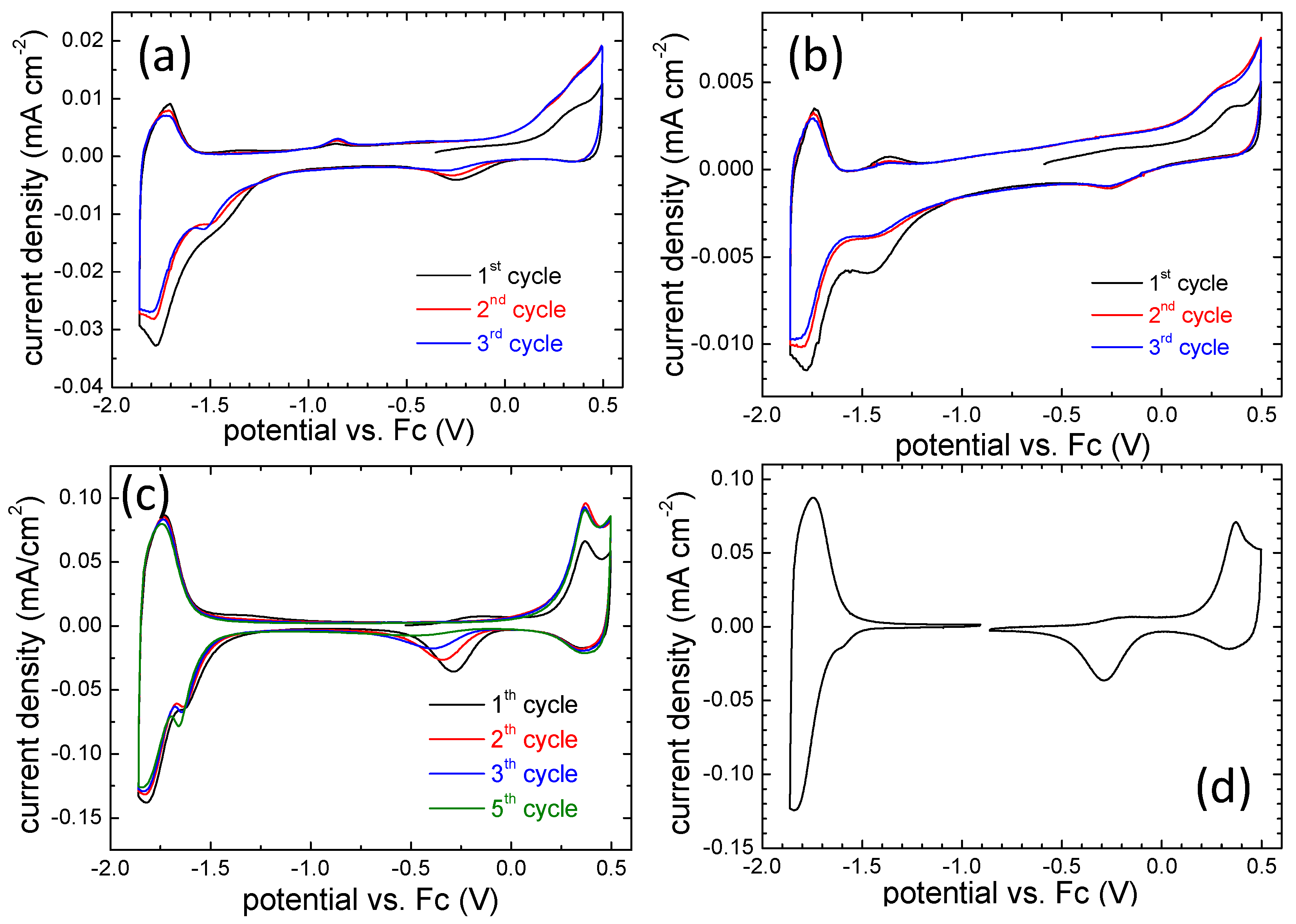
3.2. Electrochemical Behavior



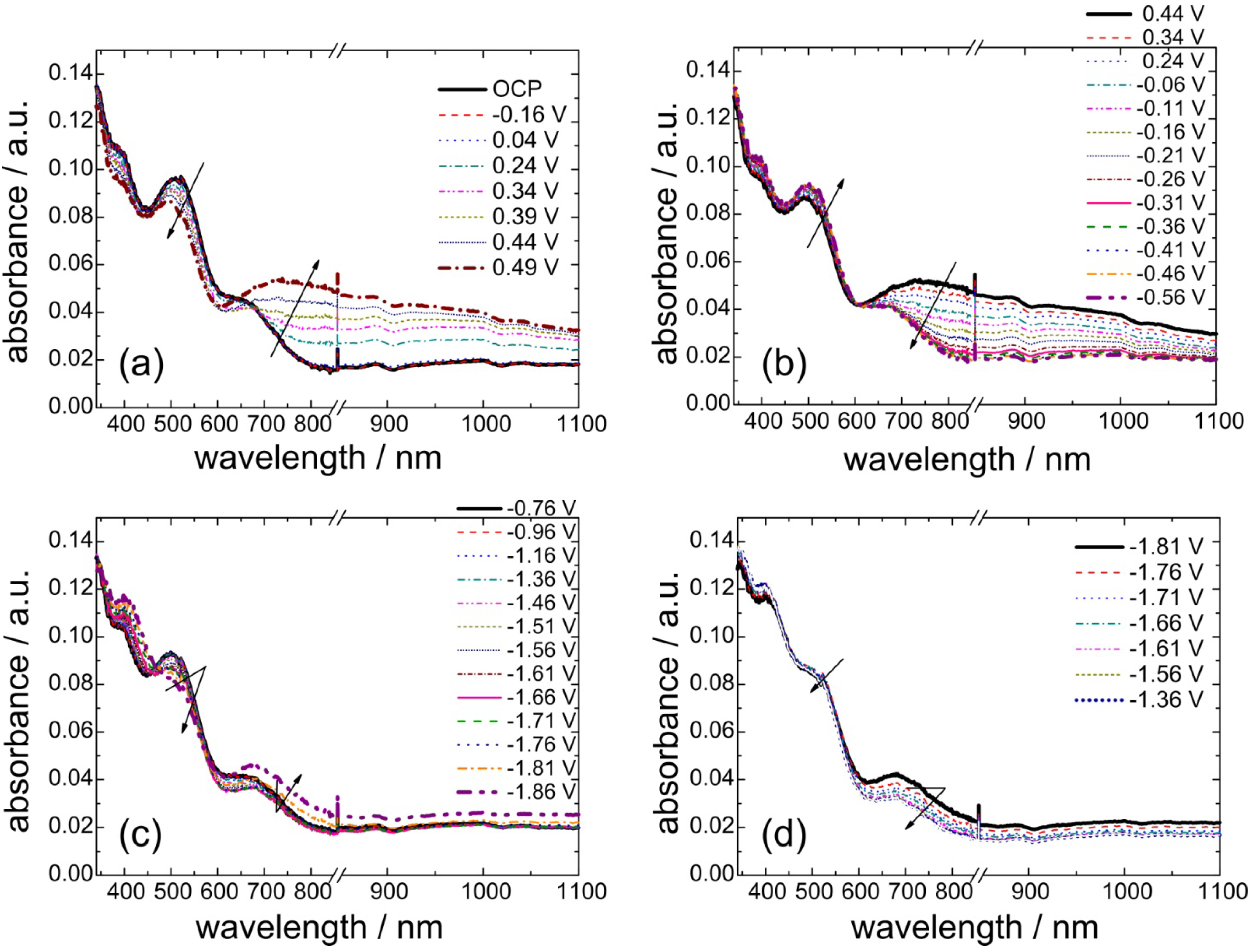
3.3. In Situ UV-Vis Spectroscopy
4. Conclusions
| Species | HOMO [eV] | LUMO [eV] | Bandgap [eV] |
|---|---|---|---|
| PCBM | −4.3 | ||
| poly(DEHT-V-BTD) | −5.3 | −3.6 | 1.7 |
| poly(2,5-pyridine-V-PEDOT) a | −5.2 | −3.2 | 2.0 |
| poly(2,5-pyridine-V-pyrrole) b | −5.4 | −3.2 | 2.2 |
Acknowledgments
Conflict of Interest
References
- Po, R.; Maggini, M.; Camaioni, N. Polymer solar cells: Recent approaches and achievements. J. Phys. Chem. C 2010, 114, 695–706. [Google Scholar]
- Havinga, E.E.; ten Hoeve, W.; Wynberg, H. A new class of small band gap organic polymer conductors. Polym. Bull. 1992, 29, 119–126. [Google Scholar]
- Brabec, C.J.; Sariciftci, N.S.; Hummelen, J.C. Plastic solar cells. Adv. Funct. Mater. 2001, 11, 15–26. [Google Scholar] [CrossRef]
- Yamamoto, T.; Zhou, Z.-H.; Kanbara, T.; Shimura, M.; Kizu, K.; Maruyama, T.; Nakamura, Y.; Fukuda, T.; Lee, B.-L.; Ooba, N.; et al. π-Conjugated donor-acceptor copolymers constituted of π-excessive and π-deficient arylene units. Optical and electrochemical properties in relation to CT structure of the polymer. J. Am. Chem. Soc. 1996, 118, 10389–10399. [Google Scholar]
- Thompson, B.C.; Kim, Y.-G.; McCarley, T.D.; Reynolds, J.R. Soluble narrow band gap and blue propylenedioxythiophene-cyanovinylene polymers as multifunctional materials for photovoltaic and electrochromic applications. J. Am. Chem. Soc. 2006, 128, 12714–12725. [Google Scholar] [CrossRef]
- Zotti, G.; Zecchin, S.; Schiavon, G.; Berlin, A.; Penso, M. Ionochromic and potentiometric properties of the novel polyconjugated polymer from anodic coupling of 5,5′-bis(3,4-(ethylenedioxy)thien-2-yl)-2,2′-bipyridine. Chem. Mater. 1999, 11, 3342–3351. [Google Scholar] [CrossRef]
- Groenendaal, L.; Zotti, G.; Aubert, P.-H.; Waybright, S.M.; Reynolds, J.R. Electrochemistry of poly(3,4-alkylenedioxythiophene) derivatives. Adv. Mater. 2003, 15, 855–879. [Google Scholar] [CrossRef]
- Beaujuge, P.M.; Ellinger, S.; Reynolds, J.R. The donor-acceptor approach allows a black-to-transmissive switching polymeric electrochrome. Nat. Mater. 2008, 7, 795–799. [Google Scholar]
- Abbotto, A.; Herrera Calderon, E.; Manfredi, N.; Mari, C.M.; Marinzi, C.; Ruffo, R. Vinylene-linked pyridine-pyrrole donor-acceptor conjugated polymers. Synth. Met. 2011, 161, 763–769. [Google Scholar] [CrossRef]
- Abbotto, A.; Calderon, E.H.; Dangate, M.S.; de Angelis, F.; Manfredi, N.; Mari, C.M.; Marinzi, C.; Mosconi, E.; Muccini, M.; Ruffo, R.; et al. Pyridine-edot heteroarylene-vinylene donor-acceptor polymers. Macromolecules 2010, 43, 9698–9713. [Google Scholar] [CrossRef]
- Roncali, J. Electrogenerated functional conjugated polymers as advanced electrode materials. J. Mater. Chem. 1999, 9, 1875–1893. [Google Scholar] [CrossRef]
- Mei, J.; Heston, N.C.; Vasilyeva, S.V.; Reynolds, J.R. A Facile Approach to defect-free vinylene-linked benzothiadiazole–thiophene low-bandgap conjugated polymers for organic electronics. Macromolecules 2009, 42, 1482–1487. [Google Scholar]
- Beaujuge, P.M.; Subbiah, J.; Choudhury, K.R.; Ellinger, S.; McCarley, T.D.; So, F.; Reynolds, J.R. Green Dioxythiophene-benzothiadiazole donor-acceptor copolymers for photovoltaic device applications. Chem. Mater. 2010, 22, 2093–2106. [Google Scholar] [CrossRef]
- King, G.; Higgins, S.J. Synthesis and characterisation of novel substituted benzo[c]thiophenes and polybenzo[c]thiophenes: Tuning the potentials for n- and p-doping in transparent conducting polymers. J. Mater. Chem. 1995, 5, 447–455. [Google Scholar] [CrossRef]
- Bard, A.J.; Faulkner, R.L. Electrochemical Methods, 2nd ed.; John Wiley and Sons: New York, NY, USA, 2001; p. 54. [Google Scholar]
- Sawyer, D.T.; Sobkowiak, A.; Roberts, J.L., Jr. Electrochemistry for Chemists, 3rd ed.; John Wiley and Sons: New York, NY, USA, 1995; p. 203. [Google Scholar]
- Schuhmann, W.; Kranz, C.; Wohlschlaeger, H.; Strohmeier, J. Pulse technique for the electrochemical deposition of polymer films on electrode surfaces. Biosens. Bioelectron. 1997, 12, 1157–1167. [Google Scholar] [CrossRef]
- Van Haare, J.A.E.H.; Havinga, E.E.; van Dongen, J.L.J.; Janssen, R.A.J.; Cornil, J.; Brèdas, J.-L. Redox states of long oligothiophenes: Two polarons on a single chain. Chem. Eur. J. 1998, 4, 1509–1522. [Google Scholar]
- Galand, E.M.; Kim, Y.-G.; Mwaura, J.K.; Jones, A.G.; McCarley, T.D.; Shrotriya, V.; Yang, Y.; Reynolds, J.R. Optimization of narrow band-gap propylenedioxythiophene:cyanovinylene copolymers for optoelectronic applications. Macromolecules 2006, 39, 9132–9142. [Google Scholar] [CrossRef]
- Beaujuge, P.M.; Vasilyeva, S.V.; Ellinger, S.; McCarley, T.D.; Reynolds, J.R. Unsaturated linkages in dioxythiophene–benzothiadiazole donor-acceptor electrochromic polymers: The key role of conformational freedom. Macromolecules 2009, 42, 3694–3706. [Google Scholar] [CrossRef]
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Calderon, E.H.; Dangate, M.; Manfredi, N.; Abbotto, A.; Salamone, M.M.; Ruffo, R.; Mari, C.M. Electrochemical and Spectroelectrochemical Properties of a New Donor–Acceptor Polymer Containing 3,4-Dialkoxythiophene and 2,1,3-Benzothiadiazole Units. Polymers 2013, 5, 1068-1080. https://doi.org/10.3390/polym5031068
Calderon EH, Dangate M, Manfredi N, Abbotto A, Salamone MM, Ruffo R, Mari CM. Electrochemical and Spectroelectrochemical Properties of a New Donor–Acceptor Polymer Containing 3,4-Dialkoxythiophene and 2,1,3-Benzothiadiazole Units. Polymers. 2013; 5(3):1068-1080. https://doi.org/10.3390/polym5031068
Chicago/Turabian StyleCalderon, Erika Herrera, Milind Dangate, Norberto Manfredi, Alessandro Abbotto, Matteo M. Salamone, Riccardo Ruffo, and Claudio M. Mari. 2013. "Electrochemical and Spectroelectrochemical Properties of a New Donor–Acceptor Polymer Containing 3,4-Dialkoxythiophene and 2,1,3-Benzothiadiazole Units" Polymers 5, no. 3: 1068-1080. https://doi.org/10.3390/polym5031068