3.1. Polymer Synthesis and Characterization
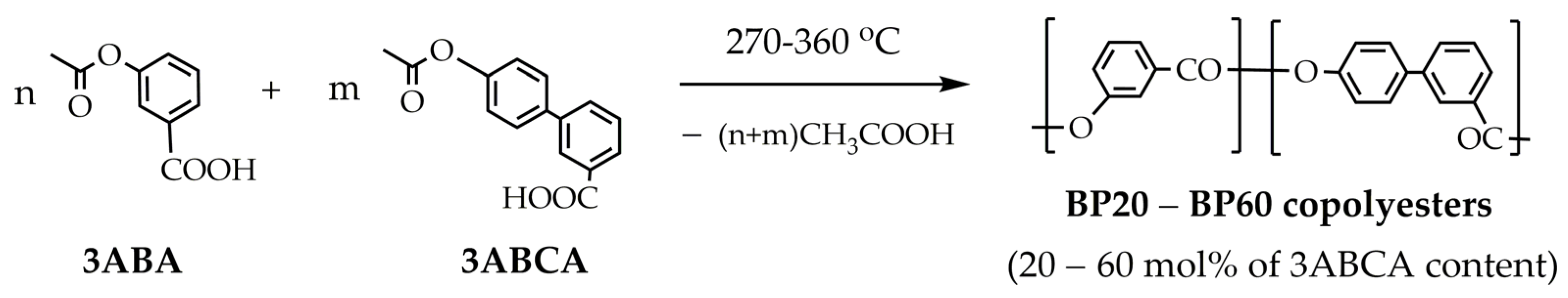
In our research, we employed the polycondensation of acetoxy derivatives of 3HBA and 3HBCA for the synthesis of copolyesters with different initial molar concentrations of 3HBCA. As mentioned above, 3HBCA is a structure similar to 3,4-biphenyldicarboxylic acid (3,4BDCA), but in contrast to 3,4BDCA, 3HBCA (and the corresponding acetyl derivative) represents an AB-type monomer; thus, it does not require the accompaniment of diol for polymer synthesis. The melt polycondensation of 3ABCA and other aromatic acetoxy acids proceeds in a single phase, because the acetoxy acids possess a low melting temperature. The scheme of the melt acidolysis polycondensation of 3ABCA and 3ABA is presented in
Figure 1.
To estimate the range of compositions that allowed for melt-processible copolyesters to be obtained, about 1 g of the 3ABCA:3ABA mixtures with variable molar ratios (100:0, 95:5, 90:10, 80:20, and 70:30) were polymerized in a 50 mL flask under the slow stream of argon. The mixture, containing 80–100% of 3ABCA, was solidified by this small-scale polymerization. Further increases in temperature up to 360 °C did not result in softening or melting. When the amount of 3HBCA decreased by up to 70%, softening was observed at about 350 °C. However, the melt was extremely viscous even at 350–360 °C, making it difficult to obtain a polymer with a sufficient molecular weight. Therefore, the amount of 3HBA was further increased and a series of polyesters with 40–80% of 3HBA was prepared (
Table 1). In addition, poly(3HBA) was prepared by the homopolycondensation of 3ABA. Polyesters containing 60 mol.% and more of 3HBA (BP40–BP20) demonstrated solubility in chloroform, unlike BP50 and BP60 which are soluble only in a chloroform/trifluoroacetic acid (TFA) mixture. All copolyesters BP60–BP20 and homopolymer poly(3HBA) were soluble in a mixture 1,2-dicholorobenzene/phenol (1:1
w/
w) and intrinsic viscosities (
IV) were measured using a Ubbelohde viscometer (values of [
η] are shown in
Table 1).
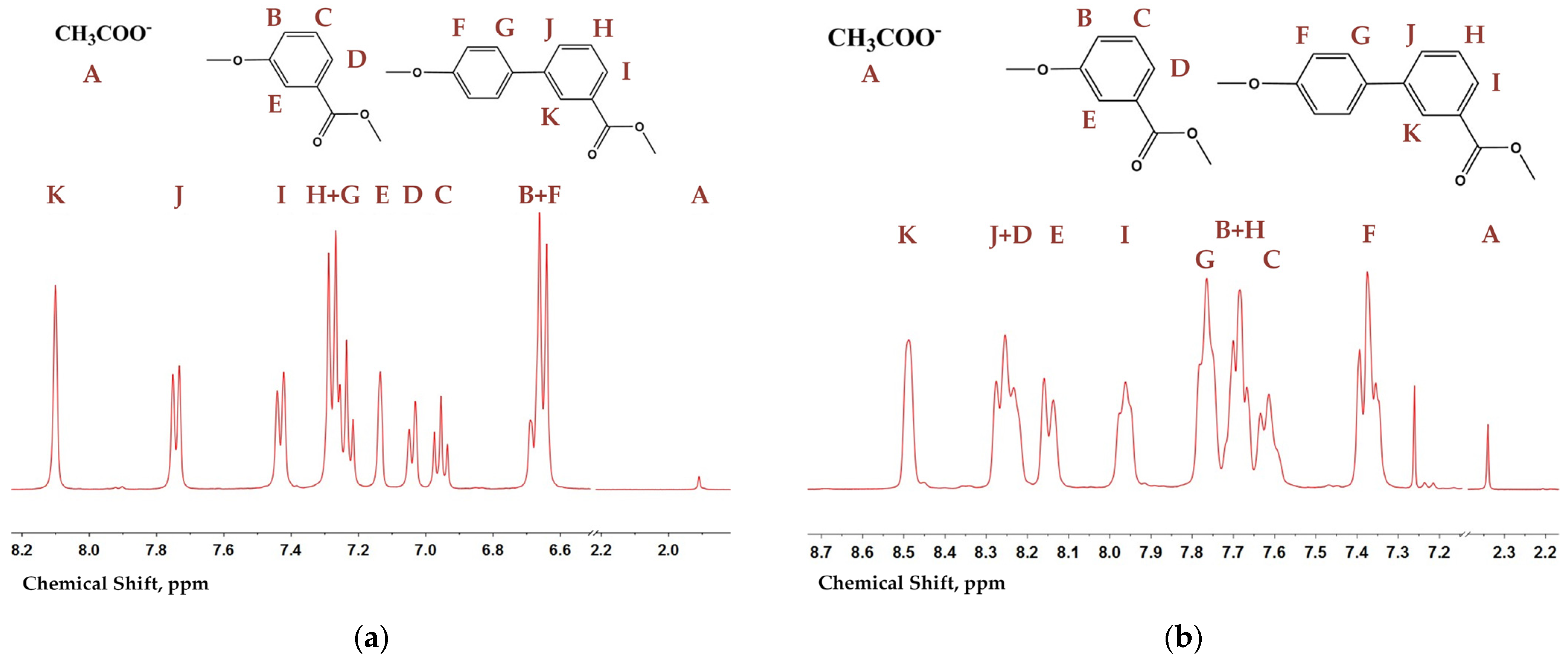
The molecular weight and composition of hydrolyzed BP60, BP40, and poly(3HBA) were determined by acetyl end group analysis performed by
1H NMR after hydrolysis in the mixture of CD
3OD/D
2O/NaOD as previously described [
22]. An example of the NMR spectra for copolyester BP50 after hydrolysis with signal assignment is shown in
Figure 2a. Signal B of 3HBA and signal F of 3HBCA overlapped, but others did not. Signal A was attributed to sodium acetate released from the acetyl end group after hydrolysis. Such spectra of copolyesters in CD
3OD/D
2O/NaOD allowed us to calculate molecular weight (
Mn) and composition (
Table 1), but did not give information about sequence distribution and randomness.
From analyzing the structure of the macromolecular backbone,
1H NMR spectra of copolyesters and poly(3HBA) in CDCl
3:CF
3COOH were obtained (example shown in
Figure 2b). The spectra of the copolyesters obtained in CDCl
3:CF
3COOH were broader in contrast to the sharp spectra obtained in CD
3OD/D
2O/NaOD, and they could not give information about the dyads and triads of monomeric units. Signal A of the acetic end group also presented in the spectra allowed for the evaluation of the composition and
Mn (
Table 1). Calculation of the composition by
1H NMR spectroscopy revealed good correlation between the targeted and obtained monomeric molar ratios with a tendency of lowering of their 3HBA content evidently due to their lower 3ABA thermal stability and higher volatility under polycondensation conditions compared with 3ABCA. The differences between the results of NMR spectroscopy of polymers in the CD
3OD/D
2O/NaOD and CDCl
3:CF
3COOH solutions were insignificant.
Molecular weights of the BP40, BP30, and BP20 samples and poly(3HBA) were measured by the GPC method using a polystyrene standard. The
Mn values obtained by GPC (
Table 1) were higher up to about 1.5 times than those obtained by
1H NMR spectroscopy. In some cases, GPC may overestimate molar weight values while NMR spectroscopy is more accurate method. Furthermore, the polydispersity index (
PDI) of the polymers increased with an increase in 3HBA content from 2.6 (BP40) to 6.3 (poly(3HBA)). On the other hand, end group analysis for copolyester prepared from acetoxy acid is suitable only for rough estimation because of side reactions [
25].
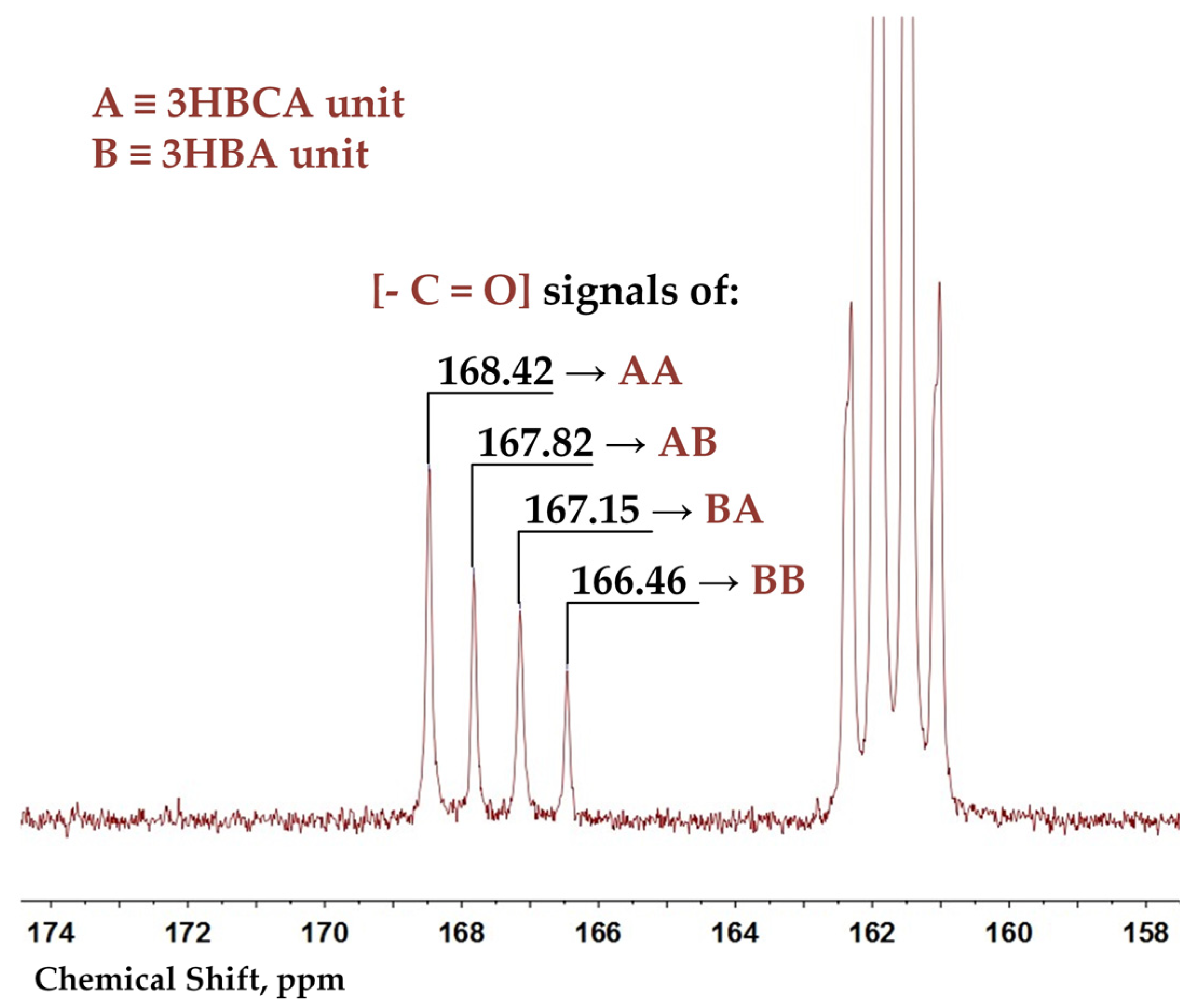
In order to distinguish triads and dyads of monomeric units in a copolyester backbone, the
13C spectra of BP40 and BP60 were recorded. Additionally, 2D COSY HSQC NMR spectra of 3-ABCA and copolyesters were recorded to make assignments (see
Supplementary Materials, Figures S1–S6). The assignments were made by the comparison of
1H,
13C, and 2D spectra of copolyesters and monomers (
Supplementary Materials, Table S1). The signals 168.31 (166.48), 167.65 (167.82), 166.99 (167.15), and 166.30 (166.46) in BP50 (BP60) spectra were assigned to the carbonyl groups of 3HBCA-3HBCA, 3HBCA-3HBA, 3HBA-3HBCA, and 3HBA-3HBA dyads, respectively (see enlarged spectrum region in
Figure 3).
The degree of randomness can be expressed by sequence order parameters according to Equations (1) and (2) [
24]:
where AA, AB, BA, and BB are the relative intensities of signals in the
13C spectra corresponding to dyads 3HBCA-3HBCA, 3HBCA-3HBA, 3HBA-3HBCA, and 3HBA-3HBA, respectively.
In the case of infinite polycondensation degrees, the number of AB and BA sequences is equal, so
ΨA =
ΨB. The value of
Ψ = 1 is valid for alternating copolyesters (ABABAB…),
Ψ = −1 for block structures (AAA…BBB…), and
Ψ = 0 for random copolyesters. Values of
ΨA and
ΨB < 0.04 for BP50 and BP60 (
Table 2) clearly demonstrate that the structure is completely random. These data correlate with the results for HBA-HNA copolyesters (
Ψ ϵ [−0.02; 0.04]) [
24] and contrast to those for the 4-oxybenzoate-1,3-phenylene terephthalate copolyester (
Ψ ϵ [−0.1; −0.02]) [
23]. The consistent trend of blockiness (
Ψ < 0) was observed for copolyesters obtained by the heteropolycondensation of the diol diacetate and dicarboxylic acid in contrast to copolyesters obtained from acetoxy acids. Remarkably, the
1H and
13C spectra gave the same values for the compositions of BP50 and BP60 (51–52% 3HBCA for BP50 and 61–62% 3HBCA for BP60).
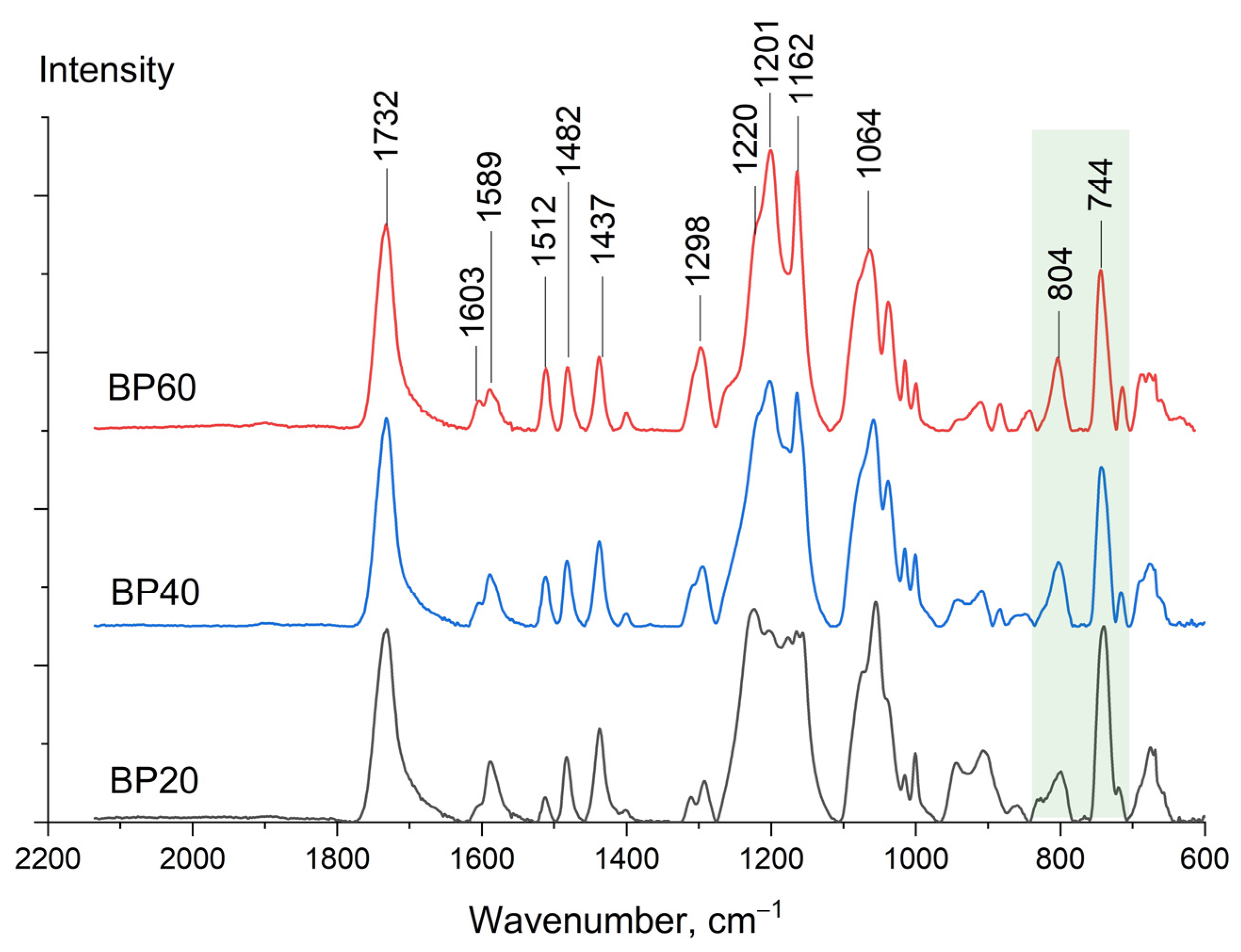
Fourier-transform infrared spectra (FTIR) were recorded for copolyesters BP60, BP40, and BP20 (
Figure 4). A band of the carbonyl group (
ν C=O) corresponded to 1732 cm
−1 and a series of benzoic ring vibrations (
ν CCH) was located in the range of 1600–1400 cm
−1. Attention should be paid to the ratio I
744/I
804 reflecting the relative ratio of 1,3- and 1,4-substituted derivatives.
Table 3 shows the ratio I
744/I
804 in the spectra normalized to the carbonyl group, as well as copolyester compositions estimated via the FTIR data. The accuracy of this estimation is not high (especially in comparison with NMR results) but it can be used to estimate composition when more sensitive methods (e.g., NMR) are unavailable.
3.2. Thermal Characteristics
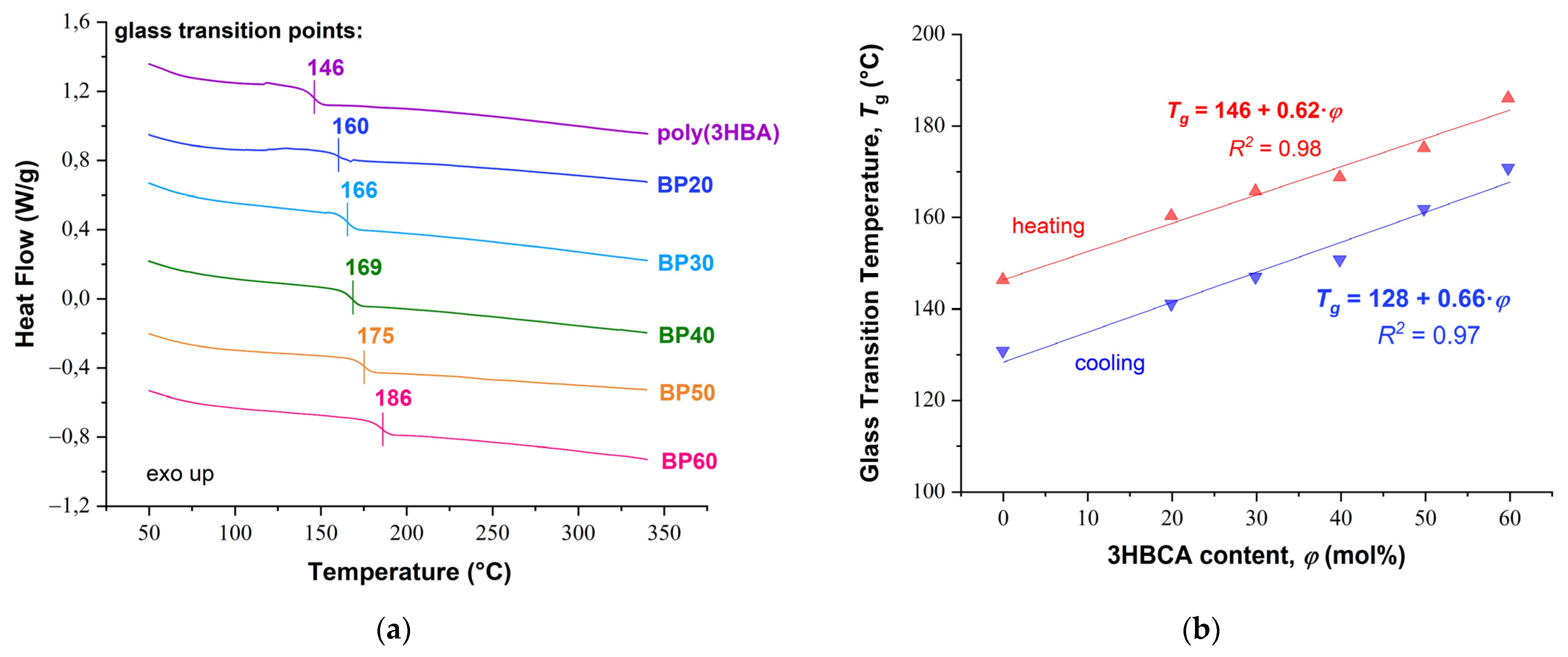
DSC thermograms for copolyesters BP20–BP60 and homopolymer poly(3HBA) are shown in
Figure 5a. The thermograms for all polymers do not demonstrate any endo- or exothermic effects that would indicate either an amorphous or mesophase structure, i.e., the absence of phase transitions in the range of 50–350 °C. At the same time, an increase in the 3HBCA content in (co)polymers resulted in significant increases in the glass transition temperature (
Tg): from 146 °C for poly(3HBA) to 186 °C for BP60. The dependence of the glass point on 3HBCA content was linear for these (co)polyesters (
Figure 5b).
Increases in the glass transition temperatures of polyesters with the introduction of biphenyl units were repeatedly mentioned earlier. The observed effect was less pronounced in the case of semi-aromatic copolyesters [
13,
20,
21]. For example, fully aromatic copolymers of 4-hydroxybenzoic acid and 4,4′-biphenol terephthalate demonstrated glass transition points up to 180 °C [
4].
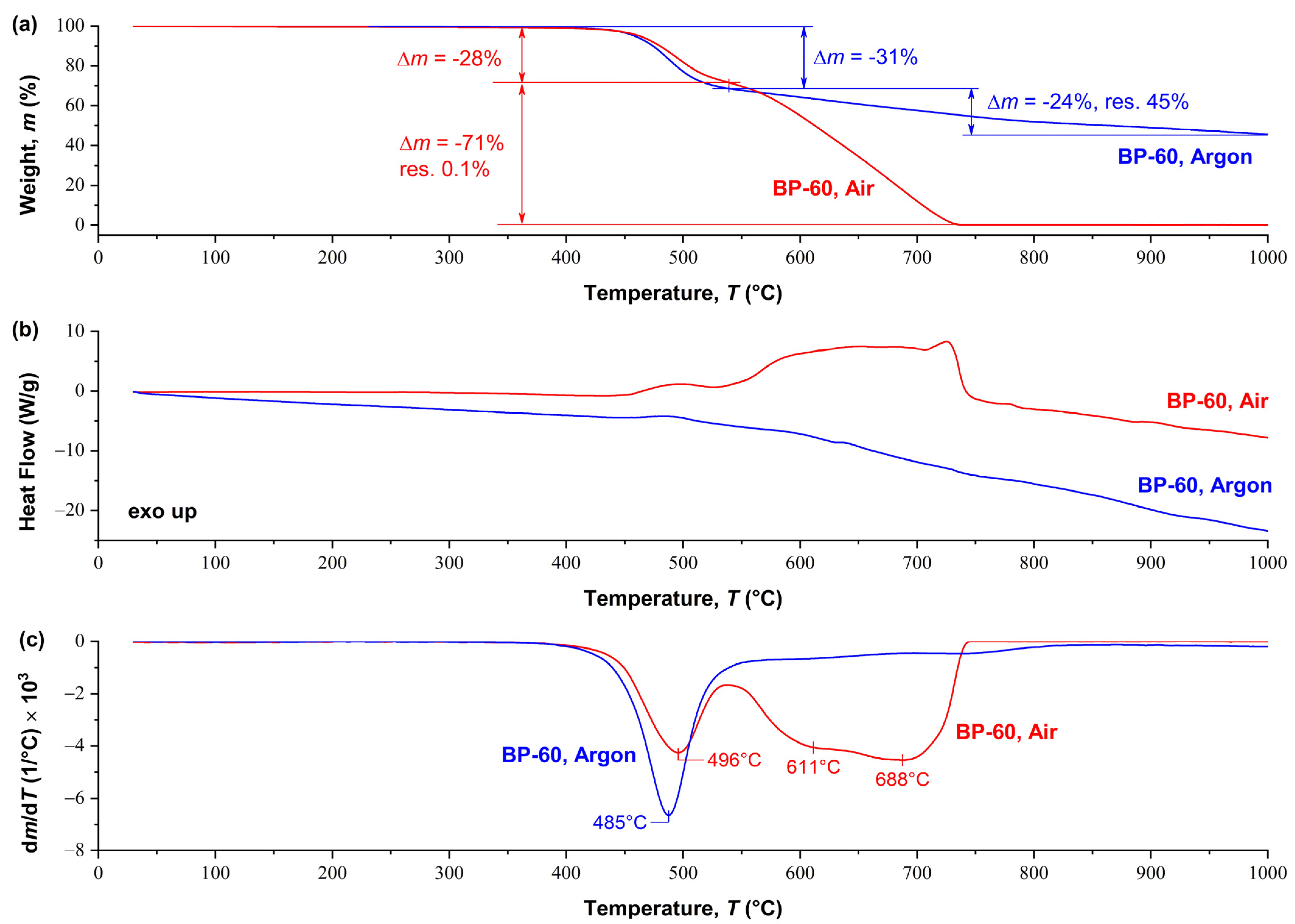
The results of synchronous TGA/DSC analysis for the BP60 copolymer in air and an inert atmosphere are shown in
Figure 6. The thermal stability of this copolyester in air and in argon is approximately the same of up to 550 °C, with mass loss at 550 °C by ~30%, 5% mass loss temperature (
TΔ5%) > 450 °C. Above 550 °C, another one-step weight loss occurs, but its magnitude and rate are radically different in air and inert media. The oxidizing atmosphere apparently accelerates the thermal degradation of the polymer, and/or supplements it with thermal oxidative processes. This leads to a loss of almost 100% of the polymer mass at ~730 °C. This process is accompanied by a sharp exothermic effect observed in the temperature range of 550–750 °C on the DSC thermogram (
Figure 6b). At the same time, heating above 550 °C in an inert atmosphere leads to a gradual loss of another 25% in weight up to 1000 °C, without any exothermic effects. For clarity,
Figure 6c shows the derivative of the thermogravimetric curve (DTG), which reflects the maximum weight loss of the sample as a function of temperature, as well as a comparison of the intensity of weight loss in inert and air media.
A similar thermal behavior was described for similar fully aromatic polyester containing 3,4-biphenylene unit based on 3,4-bibenzoic acid and hydroquinone [
16]: these copolymers show analogous values of weight loss up to 500 °C and
TΔ5%. The values of the glass points for those copolymers with a high content of bibenzoate are also about 180–190 °C.
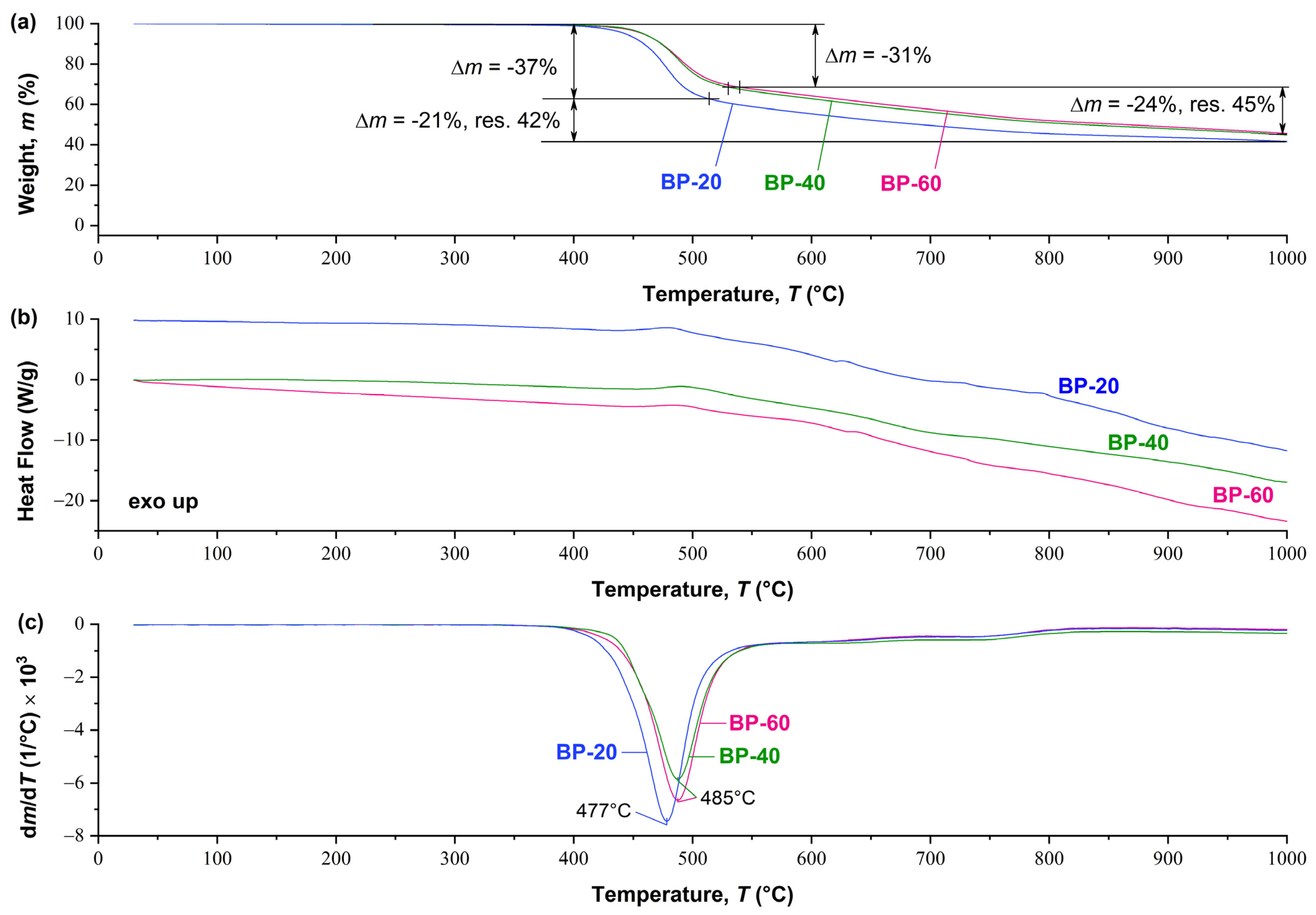
The thermal behavior of copolyesters with different content of 3HBCA units is almost identical (
Figure 7). At the same time, it should be noted that with an increase in the ratio of 3HBCA:3HBA, there is a slight increase in the thermal stability of copolyesters: the peak of maximum weight loss shifts by about +10 °C and the intensity of thermal degradation also decreases. Similar observations were previously described for ternary copolymers 4HBA-4HBCA-3HBA [
26]: the addition of a “kinked” comonomer (3HBA) led to the disappearance of thermal effects on DSC curves, as well as to a slight decrease in the thermal stability of the copolyesters. Obviously, in 3HBCA-3HBA copolyesters, a slight decrease in thermal stability upon the introduction of 3HBA is leveled by a significantly higher content of the 3HBCA biphenyl comonomer with higher thermal stability confirmed by the DSC-TGA results.
Thus, the significantly increased glass transition temperatures in combination with a sufficiently high temperature
TΔ5% allow us to state that the 3HBCA-3HBA copolyesters have a higher thermal stability not only in comparison with the semi-aromatic polyesters PET-4HBCA [
21], but also in comparison with the aromatic copolyesters 4HBCA-4HBA-3HBA [
26] obtained earlier.
3.3. Rheological Characteristics
A change in the composition of copolymers naturally affects the rheological characteristics of their melts. As previously described, the introduction of up to 80% of biphenyl units of 4HBCA into the main chain of PET leads to multiple increases in all viscoelastic properties of the copolymers [
21].
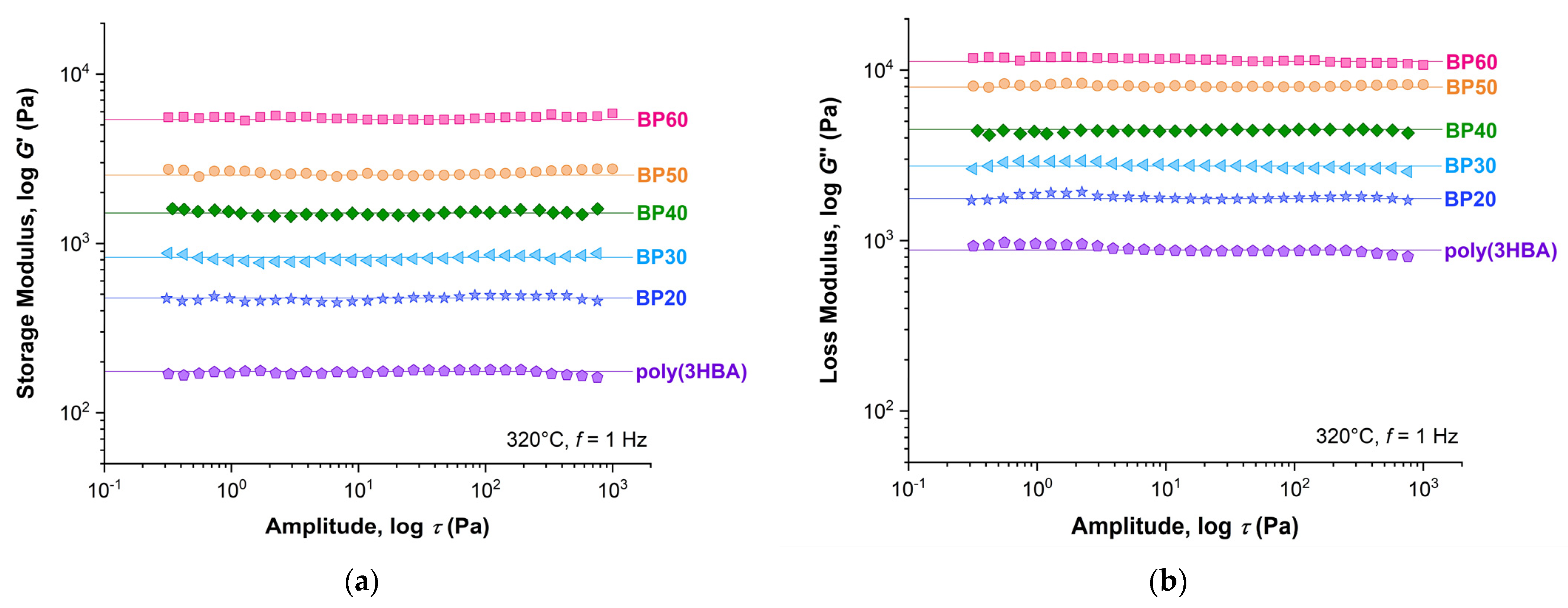
Figure 8 shows the dependence of the storage (
G′) and loss (
G″) moduli on the stress amplitude in an oscillation regime with a frequency of 1 Hz for poly(3HBA) and BP-20–BP-60 melt at 320 °C. With an increase in 3HBCA content from 20 to 60%, a significant increase in viscous and elastic properties of the melts was observed. The difference between the
G′ and
G″ values decreased in order from BP-20 to BP-60, but the viscous characteristics prevailed over elastic ones for all (co)polymers in the entire deformation range. In addition, all copolyesters exhibited a very wide range of linear viscoelasticity (LVE)—about four orders of magnitude in stress amplitude values.
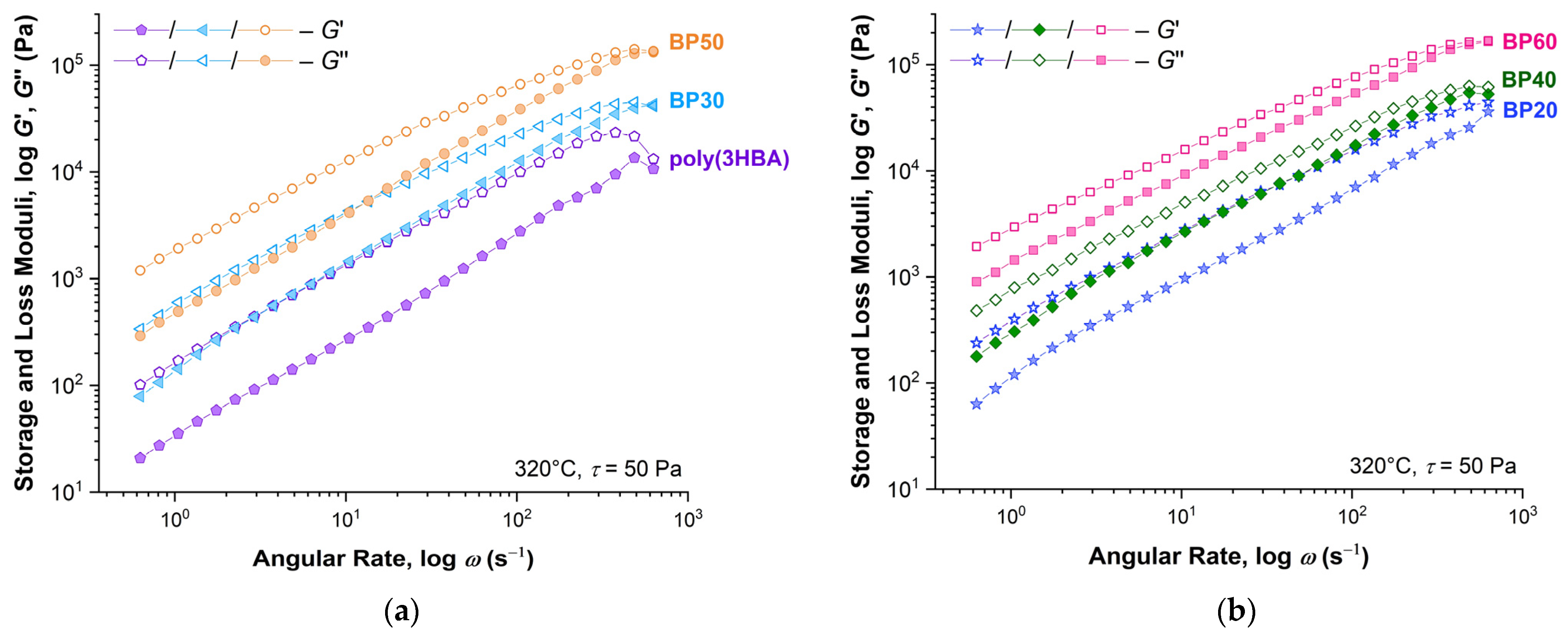
The stress value of 50 Pa was used to determine the frequency sweep of the
G′ and
G″ moduli at 320 °C, shown in
Figure 9a for samples poly(3HBA), BP30, and BP50 and in
Figure 9b for samples BP20, BP40, and BP60 (division into two figures to prevent the intersection of data series). For all samples, the values of
G″ exceeded the values of
G′ in almost the entire frequency range, which indicates a significant prevalence of the viscous characteristics over the elastic ones in polyester melts. With an increase in the oscillation frequency to 100 Hz, the values of the moduli became equal. In addition, the difference between the
G′/
G″ values decreased with an increase in the content of rigid biphenyl units of 3HBCA.
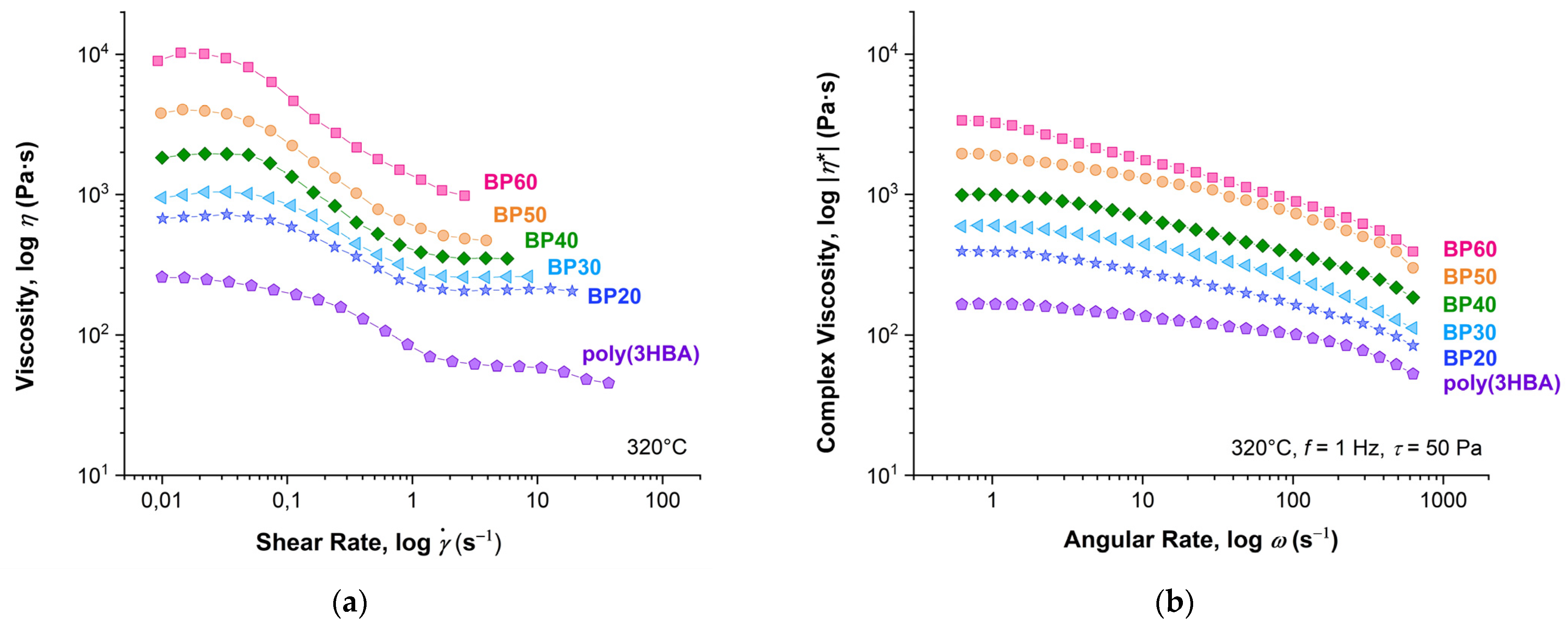
Figure 10 presents flow curves obtained in the stationary shear regime (0.01–100 s
–1) and in the oscillation mode (angular rate range 0.5–1000 s
–1) for (co)polyester melts. All samples exhibit non-Newtonian behavior. With an increase in the 3HBCA content, the viscosity values increased drastically and shear thinning became more prominent. The complex viscosity values (|
η*|) obtained by oscillation tests were at least twice as high as the shear viscosity values (
η) at the condition of equality of shear and angular rates. This means that the Cox–Merz rule [
27] does not work for these polymer melts. This can be explained by the orientational alignment in the flow, which proceeds more efficiently in shear flow for macromolecules of rigid-chain polymers. It is also worth noting that shear viscosity can only be measured up to shear rates of about 5–10 s
–1 due to the high elasticity of 3HBA:3HBCA copolyester melts. After that, the spurt effect is observed. It should also be noted that melt viscosities are more likely to correspond to the molecular weights of the copolyesters determined by NMR rather than GPC.
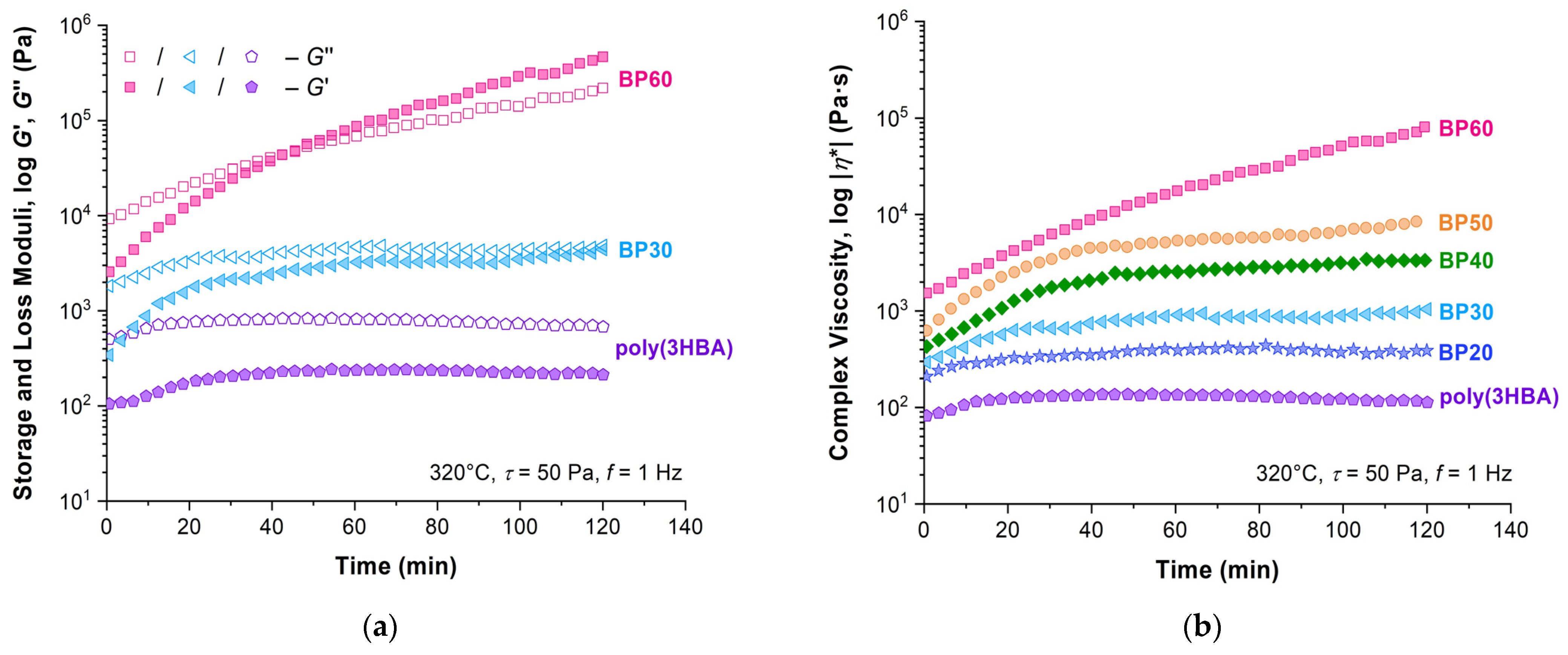
The significant increase in viscoelastic characteristics after exposure in time under a load was observed for all samples (
Figure 11). The rate of their growth and the position of the crossover point of
G′/
G″ depended on the composition being inversely proportional to the 3HBCA content. Thus, increases in moduli and complex viscosity are not significant for the poly(3HBA) homopolymer, and the intersection of the
G′/
G″ curves did not occur. With an increase in 3HBCA content, the differences between the viscosity values measured at zero-time and after 2 h increased significantly. At the same time, the crossover point was reached after an exposure of 2 h for BP30 and 50 min for BP60.
An increase in the viscoelastic characteristics in time under a constant load is not unusual for copolyesters of such structures. Similar behavior was found earlier for copolyesters of PET-4HBA [
28] and PET-4HBCA [
21]. It was shown that the elastic characteristics increased faster than the viscous ones over time, leading to the appearance of the crossover point, after which
G′ >
G″. It is remarkable that the effect of an increase in the viscoelastic characteristics in time was also observed in an inert atmosphere, which eliminated the influence of thermal oxidative processes. Moreover, the crossover transition occurred later for copolymers with a high content of mesogenic units. The observed increase in moduli may be caused by a chemical reaction in macromolecules of copolyesters of PET and 4HBA or 4HBCA caused by their presence in diethylene glycol (DEG) units [
21,
28]. The reduced amount of DEG units in the copolyester backbone with a high content of 4HBA/4HBCA causes a longer period for reaching the crossover point.
The previous explanation for modulus evolution due to degradation caused by DEG units is not suitable for fully aromatic 3HBCA-3HBA copolymers. In addition, DSC/TGA data obtained both in an inert atmosphere and in air (
Figure 6) did not demonstrate any change in polymers up to 350 °C. Moreover, the increase in 3HBCA content resulted in a higher thermal stability. A possible explanation for this consists of the reorientation of macromolecules under prolonged oscillatory loading, which occurs in melts of 3HBCA-3HBA rigid-chain copolyesters and leads to the strengthening of the reorganized internal structure. The lesser the mobility of macromolecules, i.e., with an increase in the content of 3HBCA, the longer the process. This hypothesis is supported by the fact that for less rigid-chain poly(3HBA) and for copolyesters with a low content of 3HBCA biphenyl units, the transition through the crossover was not observed or occurred noticeably later in comparison with the BP60 sample.
3.4. Polarization Optical Microscopy
In order to observe optical properties of synthesized copolyesters, their melts were analyzed by polarizing microscopy. Polymer samples (pellets), placed between two glass slides, were sequentially heated up to 375 °C (with image capture at 50, 150, 270, 320, and 375 °C) and then cooled up to 50 °C. The micrographs obtained with/without using the crossed polarizers are shown in
Figure 12.
As can be seen from the micrographs, all (co)polyesters softened after the glass transition temperature (above 150–190 °C); at 270 °C, a rather high plasticity of the melts is observed. Upon further heating and subsequent cooling, the homopolymer poly(3HBA), as well as the copolyester with a low content of biphenyl units (BP30), form homogeneous, optically transparent films. At a high content of 3HBCA fragments (sample BP60), the viscosity of the melt becomes excessively high even at 375 °C, which prevents the formation of a homogeneous film between glass slides.
For a more accurate evaluation of the melt isotropy for samples BP30 and BP60, thin films were prepared by pressing polymer powder under a load of ~5 kg at 320 °C for 1 h. Further, these films were analyzed via the same protocol (micrographs are also shown in
Figure 12). It can be noted that for BP60 at 270–320 °C, the presence of micron-sized particles with birefringence is observed in the melt. It is most probable that these particles are high-melting partially crystalline regions rather than LC phase domains. It is possible that the reason for the strong growth of modules during the long-term exposure of the BP60 melt at the temperature of the existence of these particles (
Figure 11a above) is some kind of evolution of these crystalline regions under conditions of external mechanical impact (e.g., recrystallization, growth). With further heating, these regions melt, and the sample becomes almost isotropic. After cooling, the melt forms an optically transparent glassy film with high hardness.
The obtained samples did not exhibit the optical properties inherent for nematic melts of thermotropic polyesters, as was previously shown for polymers based on 3,4BDCA-HQ (HQ—hydroquinone derivatives) [
16] and 4HBA-4HBCA-3HBA [
26]. However, in the works listed above, bent
m-substituted comonomers (3,4BDCA and 3HBA) were part of the composition in addition to
p-substituted mesogenic units. In these cases, the introduction of
m-substituted fragments expectedly reduced the crystallinity of the copolyesters, but did not prevent their transition to the nematic LC state.
In our case, the possibility that the spontaneous formation of the LC phase is likely to be suppressed for (co)polyesters was entirely formed from m-substituted rigid fragments (3HBCA and 3HBA) and block sequences were absent. Thus, it was confirmed that 3HBCA is apparently not a mesogenic monomer and neither is 3HBA.
















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}