
2,6-Diaminopyridine-Based Polyurea as an ORR Electrocatalyst of an Anion Exchange Membrane Fuel Cell


Abstract
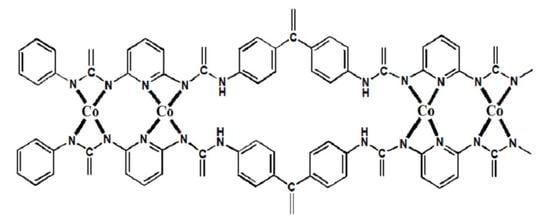
:
1. Introduction
2. Materials and Methods
2.1. Preparation
2.1.1. Preparation of Co-PUr (The Catalyst Precursor)
2.1.2. Preparation of Co–N–Cs
2.2. Characterization
2.2.1. Fourier-Transform Infrared (FTIR) Spectroscopy
2.2.2. Scanning Electron Microscopy (SEM)
2.2.3. High-Resolution Transmission Electron Microscopy (HR-TEM)
2.2.4. X-ray Diffraction Spectroscopy (XRD)
2.2.5. X-ray Photoelectronic Spectroscopy (XPS)
2.2.6. Raman Spectroscopy
2.2.7. Energy Dispersive X-ray Spectroscopy (EDs)
2.2.8. Brunauer–Emmett–Teller (BET) Analysis
2.2.9. Electrochemical Measurements
Current–Voltage (C–V) and Linear Sweep Voltammetry (LSV) Curves
Membrane Electrode Assembly (MEA)
Single Cell Testing
3. Results and Discussion
3.1. FTIR Spectra
 ), which contributes to a stretch band at ca. 1604 cm−1 in the IR spectrum of uncomplex, neat PUr, as seen in Figure 2b. After chelating with Co(II) (Co-PUr), the peak splits into two peaks at 1622 and 1604 cm−1 (demonstrating a shoulder form) following Figure 1b [39]. In other words, introducing the pyridine group into the polymer backbone can effectively increase the number of complexed Co(II).
), which contributes to a stretch band at ca. 1604 cm−1 in the IR spectrum of uncomplex, neat PUr, as seen in Figure 2b. After chelating with Co(II) (Co-PUr), the peak splits into two peaks at 1622 and 1604 cm−1 (demonstrating a shoulder form) following Figure 1b [39]. In other words, introducing the pyridine group into the polymer backbone can effectively increase the number of complexed Co(II).3.2. SEM Micropictures
3.3. HR-TEM Micropictures
3.4. XRD Pattern
3.5. XPS
3.6. Raman Spectroscopy
3.7. EDs
3.8. BET Surface Area and Pore Size
3.9. Electrochemical Measurements
3.9.1. C–V and LSV Curves
3.9.2. Single-Cell Testing
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Zhang, Y.; Pan, Q.; Chai, G.; Liang, M.; Dong, G.; Zhang, Q.; Qiu, J. Synthesis and luminescence mechanism of multicolor-emitting g-C3N4 nanopowders by low temperature thermal condensation of melamine. Sci. Rep. 2013, 3, 1943. [Google Scholar] [CrossRef]
- Rahbar, N.; Salehnezhad, Z.; Hatamie, A.; Babapour, A. Graphitic carbon nitride nanosheets as a fluorescent probe for chromium speciation. Microchim. Acta 2018, 185, 101. [Google Scholar] [CrossRef]
- Sun, S.; Gou, X.; Tao, S.; Cui, J.; Li, J.; Yang, Q.; Liang, S.; Yang, Z. Mesoporous graphitic carbon nitride (g-C3N4) nanosheets synthesized from carbonated beverage-reformed commercial melamine for enhanced photocatalytic hydrogen evolution. Mater. Chem. Front. 2019, 3, 597–605. [Google Scholar] [CrossRef]
- Zhong, H.; Estudillo-Wong, L.A.; Gao, Y.; Feng, Y.; Alonso-Vante, N. Cobalt-Based Multicomponent Oxygen Reduction Reaction Electrocatalysts Generated by Melamine Thermal Pyrolysis with High Performance in an Alkaline Hydrogen/Oxygen Microfuel Cell. ACS Appl. Mater. Interfaces 2020, 12, 21605–21615. [Google Scholar] [CrossRef] [PubMed]
- Peng, H.; Liu, F.; Liu, X.; Liao, S.; You, C.; Tian, X.; Nan, H.; Luo, F.; Song, H.; Fu, Z.; et al. Effect of Transition Metals on the Structure and Performance of the Doped Carbon Catalysts Derived from Polyaniline and Melamine for ORR Application. ACS Catal. 2014, 4, 3797–3805. [Google Scholar] [CrossRef]
- Sheng, Z.-H.; Shao, L.; Chen, J.-J.; Bao, W.-J.; Wang, F.-B.; Xia, X.-H. Catalyst-Free Synthesis of Nitrogen-Doped Graphene via Thermal Annealing Graphite Oxide with Melamine and Its Excellent Electrocatalysis. ACS Nano 2011, 5, 4350–4358. [Google Scholar] [CrossRef] [PubMed]
- Lee, W.H.; Lee, D.W.; Kim, H. Development of Nitrogen-Doped Carbon Catalysts Using Melamine-Based Polymer as a Nitrogen Precursor for the Oxygen Reduction Reaction. J. Electrochem. Soc. 2015, 162, F744. [Google Scholar] [CrossRef]
- Xia, D.; Wang, R.; Wei, Y.; Gan, L.; Kang, F. Melamine-sponge-derived non-precious fuel cell electrocatalyst with hierarchical pores and tunable nitrogen chemical states for exceptional oxygen reduction reaction activity. Mater. Today Energy 2018, 9, 271–278. [Google Scholar] [CrossRef]
- Aghabarari, B.; Martínez-Huerta, M.V.; Capel-Sánchez, M.C.; Lázaro, M.J. Non-precious Melamine/Chitosan Composites for the Oxygen Reduction Reaction: Effect of the Transition Metal. Front. Mater. 2020, 7, 578518. [Google Scholar] [CrossRef]
- Zhang, C.; Zhang, W.; Zheng, W. Transition Metal-Nitrogen-Carbon Active Site for Oxygen Reduction Electrocatalysis: Beyond the Fascinations of TM-N4. ChemCatChem 2019, 11, 655–668. [Google Scholar] [CrossRef]
- Marshall-Roth, T.; Libretto, N.J.; Wrobel, A.T.; Anderton, K.J.; Pegis, M.L.; Ricke, N.D.; Voorhis, T.V.; Miller, J.T.; Surendranath, Y. A pyridinic Fe-N4 macrocycle models the active sites in Fe/N-doped carbon electrocatalysts. Nat. Commun. 2020, 11, 5283. [Google Scholar] [CrossRef] [PubMed]
- Asset, T.; Atanassov, P. Iron-Nitrogen-Carbon Catalysts for Proton Exchange Membrane Fuel Cells. Joule 2020, 4, 33–44. [Google Scholar] [CrossRef]
- Mehmood, A.; Gong, M.; Jaouen, F.; Roy, A.; Zitolo, A.; Khan, A.; Sougrati, M.-T.; Primbs, M.; Bonastre, A.M.; Fongalland, D.; et al. High loading of single atomic iron sites in Fe–NC oxygen reduction catalysts for proton exchange membrane fuel cells. Nat. Catal. 2022, 5, 311–323. [Google Scholar] [CrossRef]
- Wang, M.; Huang, B.; Jiang, N.; Liu, T.; Huang, J.; Guan, L. An Fe–N–C electrocatalyst with dense active sites synthesized by expeditious pyrolysis of a natural Fe–N4 macrocyclic complex. J. Mater. Chem. A 2022, 10, 23001–23007. [Google Scholar] [CrossRef]
- Tian, Z.; Wang, Y.; Li, Y.; Yao, G.; Zhang, Q.; Chen, L. Theoretical study of the effect of coordination environment on the activity of metal macrocyclic complexes as electrocatalysts for oxygen reduction. iScience 2022, 25, 104557. [Google Scholar] [CrossRef]
- Pegis, M.L.; Wise, C.F.; Martin, D.J.; Mayer, J.M. Oxygen Reduction by Homogeneous Molecular Catalysts and Electrocatalysts. Chem. Rev. 2018, 118, 2340–2391. [Google Scholar] [CrossRef]
- Pegis, M.L.; Martin, D.J.; Wise, C.F.; Brezny, A.C.; Johnson, S.I.; Johnson, L.E.; Kumar, N.; Raugei, S.; Mayer, J.M. Mechanism of Catalytic O2 Reduction by Iron Tetraphenylporphyrin. J. Am. Chem. Soc. 2019, 141, 8315–8326. [Google Scholar] [CrossRef]
- Liang, Z.; Wang, H.-Y.; Zheng, H.; Zhang, W.; Cao, R. Porphyrin-based frameworks for oxygen electrocatalysis and catalytic reduction of carbon dioxide. Chem. Soc. Rev. 2021, 50, 2540–2581. [Google Scholar] [CrossRef]
- Han, X.; Wang, Y.; Zhang, Y.; Cong, Y.; Qin, J.; Gao, R.; Chai, C.; Song, Y. Oxygen Reduction Reaction Electrocatalysts Derived from Metalloporphyrin-Modified Meso-/Macroporous Polyaniline. Acta Phys.-Chim. Sin. 2021, 37, 2008017. [Google Scholar] [CrossRef]
- Yao, B.; He, Y.; Wang, S.; Sun, H.; Liu, X. Recent Advances in Porphyrin-Based Systems for Electrochemical Oxygen Evolution Reaction. Int. J. Mol. Sci. 2022, 23, 6036. [Google Scholar] [CrossRef]
- Orellana, W.; Loyola, C.Z.; Marco, J.F.; Tasca, F. Evidence of carbon-supported porphyrins pyrolyzed for the oxygen reduction reaction keeping integrity. Sci. Rep. 2022, 12, 8072. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Zhang, S.; Wang, Y.; Si, J.; Chen, Y.; Zhuang, L.; Chen, S. Boosting the Performance of Iron-Phthalocyanine as Cathode Electrocatalyst for Alkaline Polymer Fuel Cells Through Edge-Closed Conjugation. ACS Appl. Mater. Interfaces 2018, 10, 28664–28671. [Google Scholar] [CrossRef] [PubMed]
- Viera, M.; Riquelme, J.; Aliaga, C.; Marco, J.F.; Orellana, W.; Zagal, J.H.; Tasca, F. Oxygen Reduction Reaction at Penta-Coordinated Co Phthalocyanines. Front. Chem. 2020, 8, 22. [Google Scholar] [CrossRef]
- Park, J.S.; Chang, D.W. Iron Phthalocyanine/Graphene Composites as Promising Electrocatalysts for the Oxygen Reduction Reaction. Energies 2020, 13, 4073. [Google Scholar] [CrossRef]
- Yoon, J.; Kim, S.; Park, H.; Prabakar, K.; Lun Li, O. Molecular M-N4 macrocycles in a nitrogen-carbon matrix as a highly durable oxygen reduction reaction (ORR) electrocatalysts in acid media. Mater. Lett. 2021, 291, 129561. [Google Scholar] [CrossRef]
- Yuan, S.; Peng, J.; Zhang, Y.; Zheng, D.J.; Bagi, S.; Wang, T.; Román-Leshkov, Y.; Shao-Horn, Y. Tuning the Catalytic Activity of Fe-Phthalocyanine-Based Catalysts for the Oxygen Reduction Reaction by Ligand Functionalization. ACS Catal. 2022, 12, 7278–7287. [Google Scholar] [CrossRef]
- Li, X.; Wang, Z.; Su, Z.; Zhao, Z.; Cai, Q.; Zhao, J. Phthalocyanine-supported single-atom catalysts as a promising bifunctional electrocatalyst for ORR/OER: A computational study. ChemPhysMater 2022, 1, 237–245. [Google Scholar] [CrossRef]
- Zhang, W.; Meeus, E.J.; Wang, L.; Zhang, L.-H.; Yang, S.; de Bruin, B.; Reek, J.N.H.; Yu, F. Boosting Electrochemical Oxygen Reduction Performance of Iron Phthalocyanine through Axial Coordination Sphere Interaction. ChemSusChem 2022, 15, e202102379. [Google Scholar] [CrossRef]
- Chen, S.; Xu, Z.; Li, J.; Yang, J.; Shen, X.; Zhang, Z.; Li, H.; Li, W.; Li, Z. Nanostructured transition-metal phthalocyanine complexes for catalytic oxygen reduction reaction. Nanotechnology 2022, 33, 182001. [Google Scholar] [CrossRef]
- Huang, W.Y.; Jheng, L.C.; Hsieh, T.H.; Ho, K.S.; Wang, Y.Z.; Gao, Y.J.; Tseng, P.H. Calcined Co(II)-Triethylenetetramine, Co(II)- Polyaniline-Thiourea as the Cathode Catalyst of Proton Exchanged Membrane Fuel Cell. Polymers 2020, 12, 3070. [Google Scholar] [CrossRef] [PubMed]
- Hsieh, T.H.; Chen, S.N.; Wang, Y.Z.; Ho, K.S.; Chuang, J.K.; Ho, L.C. Cobalt-Doped Carbon Nitride Frameworks Obtained from Calcined Aromatic Polyimines as Cathode Catalyst of Anion Exchange Membrane Fuel Cells. Membranes 2022, 12, 74. [Google Scholar] [CrossRef] [PubMed]
- Cheng, Y.W.; Hsieh, T.H.; Huang, Y.C.; Tseng, P.H.; Wang, Y.Z.; Ho, K.S.; Huang, Y.J. Calcined Co(II)-Chelated Polyazomethine as Cathode Catalyst of Anion Exchange Membrane Fuel Cells. Polymers 2022, 14, 1784. [Google Scholar] [CrossRef]
- Hsieh, T.H.; Wang, Y.Z.; Ho, K.S. Cobalt-Based Cathode Catalysts for Oxygen-Reduction Reaction in an Anion Exchange Membrane Fuel Cell. Membranes 2022, 12, 699. [Google Scholar] [CrossRef] [PubMed]
- Kojidi, M.H.; Aliakbar, A. A graphene oxide based poly(2,6-diaminopyridine) composite for solid-phase extraction of Cd(II) prior to its determination by FAAS. Microchim. Acta 2017, 184, 2855–2860. [Google Scholar] [CrossRef]
- Brivary, M.A.; Gómez, M.; Iglesias, M.; Maya, E.M. Accessible microwave synthetized conjugated poly(azomethine-pyridine) network and its metal complexes for CO2 conversion. J. Polym. Sci. Part A Polym. Chem. 2018, 56, 1946–1952. [Google Scholar] [CrossRef]
- Maya, E.M.; Verde-Sesto, E.; Mantione, D.; Iglesias, M.; Mecerreyes, D. New poly(ionic liquid)s based on poly(azomethine-pyridinium) salts and its use as heterogeneous catalysts for CO2 conversion. Eur. Polym. J. 2019, 110, 107–113. [Google Scholar] [CrossRef]
- Roy, N.; Yasmin, S.; Jeon, S. Effective electrochemical detection of dopamine with highly active molybdenum oxide nanoparticles decorated on 2,6 diaminopyridine/reduced graphene oxide. Microchem. J. 2020, 153, 104501. [Google Scholar] [CrossRef]
- Liu, Z.; Liu, Q.; Dai, X.; Shen-Tu, C.; Yao, C.; Kong, Y. Synthesis of Poly(2,6-diaminopyridine) Using Interface Polymerization and the Electrochemical Properties of Poly(2,6-diaminopyridine). ECS Electrochem. Lett. 2013, 2, G1. [Google Scholar] [CrossRef]
- Hansen, G.P. (Austin, TX, US); Dominguez, R.J.G. (Austin, TX, US); Hoppens, N.C. (Austin, TX, US); Shields, E.S. (Austin, TX, US); Bulluck, J.W. (Spicewood, TX, US); Rushing, R.A. (Spicewood, TX, US). Novel Polyurea Fiber. US Patent: 20110082274A1, 2011. Available online: https://www.freepatentsonline.com/y2011/0082274.html (accessed on 7 April 2011).
- Wang, X.X.; Prabhakaran, V.; He, Y.; Shao, Y.; Wu, G. Iron-Free Cathode Catalysts for Proton-Exchange-Membrane Fuel Cells: Cobalt Catalysts and the Peroxide Mitigation Approach. Adv. Mater. 2019, 31, 1805126. [Google Scholar] [CrossRef]
- Lin, M.; Tan, J.P.Y.; Boothroyd, C.; Loh, K.P.; Tok, E.S.; Foo, Y.-L. Dynamical Observation of Bamboo-like Carbon Nanotube Growth. Nano Lett. 2007, 7, 2234–2238. [Google Scholar] [CrossRef]
- Chen, Z.; Higgins, D.; Tao, H.; Hsu, R.S.; Chen, Z. Highly Active Nitrogen-Doped Carbon Nanotubes for Oxygen Reduction Reaction in Fuel Cell Applications. J. Phys. Chem. C 2009, 113, 21008–21013. [Google Scholar] [CrossRef]
- He, Z.; Maurice, J.-L.; Gohier, A.; Lee, C.S.; Pribat, D.; Cojocaru, C.S. Iron Catalysts for the Growth of Carbon Nanofibers: Fe, Fe3C or Both? Chem. Mater. 2011, 23, 5379–5387. [Google Scholar] [CrossRef]
- Fan, W.; Li, Z.; You, C.; Zong, X.; Tian, X.; Miao, S.; Shu, T.; Li, C.; Liao, S. Binary Fe, Cu-doped bamboo-like carbon nanotubes as efficient catalyst for the oxygen reduction reaction. Nano Energy 2017, 37, 187–194. [Google Scholar] [CrossRef]
- Povie, G.; Segawa, Y.; Nishihara, T.; Miyauchi, Y.; Itami, K. Synthesis of a carbon nanobelt. Science 2017, 356, 172. [Google Scholar] [CrossRef]
- Chen, H.; Gui, S.; Zhang, Y.; Liu, Z.; Miao, Q. Synthesis of a Hydrogenated Zigzag Carbon Nanobelt. CCS Chem. 2021, 3, 613–619. [Google Scholar] [CrossRef]
- Zhang, J.; Pei, Y.; Zhu, W.; Liu, Y.; Yin, Y.; Qin, Y.; Guiver, M.D. Ionomer dispersion solvent influence on the microstructure of Co–N–C catalyst layers for anion exchange membrane fuel cell. J. Power Sources 2021, 484, 229259. [Google Scholar] [CrossRef]
- Lilloja, J.; Kibena-Poldsepp, E.; Sarapuu, A.; Kikas, A.; Kisand, V.; Kaarik, M.; Merisalu, M.; Treshchalov, A.; Leis, J.; Sammelselg, V.; et al. Nitrogen-doped carbide-derived carbon/carbon nanotube composites as cathode catalysts for anion exchange membrane fuel cell application. Appl. Catal. B Environ. 2020, 272, 119012. [Google Scholar] [CrossRef]
- Zhang, J.F.; Zhu, W.K.; Pei, Y.B.; Liu, Y.; Qin, Y.Z.; Zhang, X.W.; Wang, Q.F.; Yin, Y.; Guiver, M.D. Hierarchically Porous Co-N-C Cathode Catalyst Layers for Anion Exchange Membrane Fuel Cells. Chemsuschem 2019, 12, 4165–4169. [Google Scholar] [CrossRef]
- Ratso, S.; Kruusenberg, I.; Käärik, M.; Kook, M.; Puust, L.; Saar, R.; Leis, J.; Tammeveski, K. Highly efficient transition metal and nitrogen co-doped carbide-derived carbon electrocatalysts for anion exchange membrane fuel cells. J. Power Sources 2018, 375, 233–243. [Google Scholar] [CrossRef]
- Kruusenberg, I.; Ramani, D.; Ratso, S.; Joost, U.; Saar, R.; Rauwel, P.; Kannan, A.M.; Tammeveski, K. Cobalt–Nitrogen Co-doped Carbon Nanotube Cathode Catalyst for Alkaline Membrane Fuel Cells. ChemElectroChem 2016, 3, 1455–1465. [Google Scholar] [CrossRef]
- Li, X.G.; Popov, B.N.; Kawahara, T.; Yanagi, H. Non-precious metal catalysts synthesized from precursors of carbon, nitrogen, and transition metal for oxygen reduction in alkaline fuel cells. J. Power Sources 2011, 196, 1717–1722. [Google Scholar] [CrossRef]
- Wang, C.H.; Yang, C.W.; Lin, Y.C.; Chang, S.T.; Chang, S.L.Y. Cobalt-iron(II,III) oxide hybrid catalysis with enhanced catalytic activities for oxygen reduction in anion exchange membrane fuel cell. J. Power Sources 2015, 277, 147–154. [Google Scholar] [CrossRef]
- Kisand, K.; Sarapuu, A.; Danilian, D.; Kikas, A.; Kisand, V.; Rahn, M.; Treshchalov, A.; Kaarik, M.; Merisalu, M.; Paiste, P.; et al. Transition metal-containing nitrogen-doped nanocarbon catalysts derived from 5-methylresorcinol for anion exchange membrane fuel cell application. J. Colloid Interface Sci. 2021, 584, 263–274. [Google Scholar] [CrossRef] [PubMed]
- Peng, X.; Kashyap, V.; Ng, B.; Kurungot, S.; Wang, L.Q.; Varcoe, J.R.; Mustain, W.E. High-Performing PGM-Free AEMFC Cathodes from Carbon-Supported Cobalt Ferrite Nanoparticles. Catalysts 2019, 9, 264. [Google Scholar] [CrossRef] [Green Version]












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| CoNC- | ID/IG a |
|---|---|
| 700A600 | 1.04 |
| 800A700 | 1.00 |
| 900A800 | 0.99 |
| CoNC- | C | N | O | Co |
|---|---|---|---|---|
| 700A600 | 67.10 | 19.80 | 12.21 | 0.80 |
| 800A700 | 83.80 | 8.12 | 6.85 | 1.23 |
| 900A800 | 83.80 | 9.30 | 5.10 | 1.80 |
| Co–N–Cs | BET Surface Area (m2g−1) a | BJH Adsorption Average Pore Diameter (nm) b |
|---|---|---|
| 700A600 | 177.74 | 4.17 |
| 800A700 | 369.43 | 5.24 |
| 900A800 | 447.81 | 6.18 |
| Catalysts (Pt/C & CoNC-) | On-Set Voltage (V) a | Half-Wave Voltage (V) b | LRCD (mAcm−2) c | Tafel Slope (mVdec−1) d | # of e-Transferred e | Pmax f |
|---|---|---|---|---|---|---|
| 700A600 | 0.78 | 0.39 | −2.58 | 164.1 | 2.5 | 35.8 |
| 800A700 | 0.85 | 0.41 | −3.02 | 165.9 | 3.5 | 67.0 |
| 900A800 | 0.88 | 0.82 | −5.10 | 66.2 | 4.0 | 227.7 |
| Pt/C | 0.89 | 0.83 | −5.26 | 67.9 | - | 285.3 |
| Cathode Catalyst | Cathode Loading (mgcm−2) | Anode Catalyst | Power Density (mWcm−2) | Ref. |
|---|---|---|---|---|
| Co–N–C | 0.8 | 20 wt% Pt/C | 228 | This work |
| Pt/C | 0.8 | 20 wt% Pt/C | 285 | This work |
| Co–N–C | 2.0 | 60 wt% Pt/C | 182 | [47] |
| N-dopes CDC/CNT | 0.4 | 40 wt% Pt/C | 310 | [48] |
| ZIF-CB-700 | 0.4 | 60 wt% Pt/C | 122 | [49] |
| Co/N/MWCNT | 0.6 | 40 wt% Pt/C | 115 | [50] |
| Co/N/MWCNT | 0.6 | 40 wt% Pt/C | 115 | [50] |
| Co/N/CDC | 0.8 | 60 wt% Pt/C | 78 | [51] |
| Co-Fe3O4/C | 0.8 | Pt/C | 114 | [52] |
| Co–N–C | 0.4 | 60% Pt/C | 181 | [53] |
| Co–N–CDC/CNT | 0.4 | Pt-Ru/C (wt% 50/25/25) | 577 | [47] |
| FeCoNC-at | 2.0 | Pt-Ru/C (wt% 50/25/25) | 415 | [54] |
| CoFe2O4 on Vulcan XC-72 | 2.4 | Pt-Ru | 670 | [55] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, Y.-Z.; Hsieh, T.-H.; Huang, Y.-C.; Ho, K.-S. 2,6-Diaminopyridine-Based Polyurea as an ORR Electrocatalyst of an Anion Exchange Membrane Fuel Cell. Polymers 2023, 15, 915. https://doi.org/10.3390/polym15040915
Wang Y-Z, Hsieh T-H, Huang Y-C, Ho K-S. 2,6-Diaminopyridine-Based Polyurea as an ORR Electrocatalyst of an Anion Exchange Membrane Fuel Cell. Polymers. 2023; 15(4):915. https://doi.org/10.3390/polym15040915
Chicago/Turabian StyleWang, Yen-Zen, Tar-Hwa Hsieh, Yu-Chang Huang, and Ko-Shan Ho. 2023. "2,6-Diaminopyridine-Based Polyurea as an ORR Electrocatalyst of an Anion Exchange Membrane Fuel Cell" Polymers 15, no. 4: 915. https://doi.org/10.3390/polym15040915