
Simultaneous Enhancement of the Mechanical Properties, Performance and Insensitivity of an Energetic Elastomeric Polyurethane Binder by Kinetically Grafting Reactive Spiranes
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results and Discussion
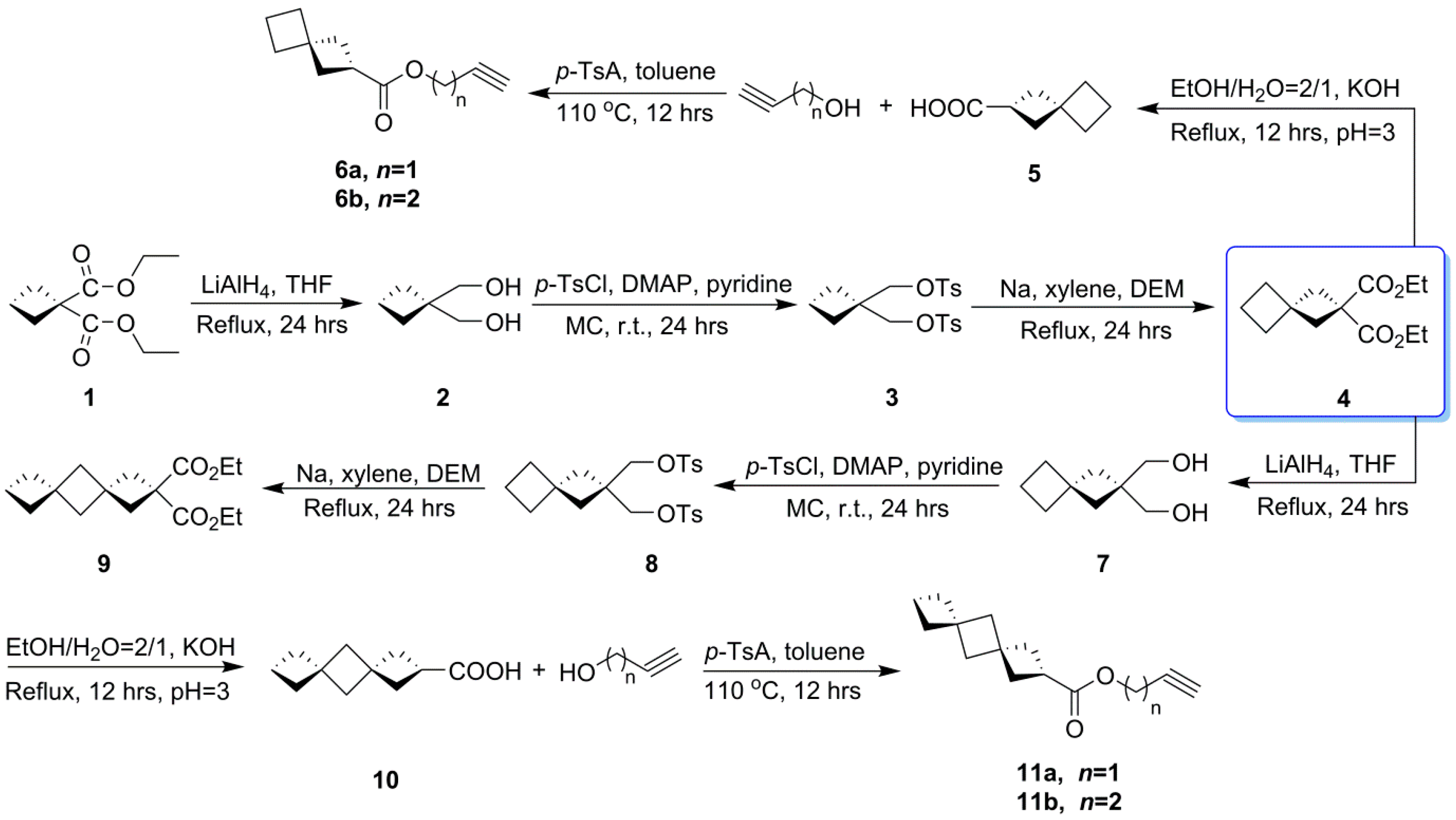
2.1. Synthesis of RGS
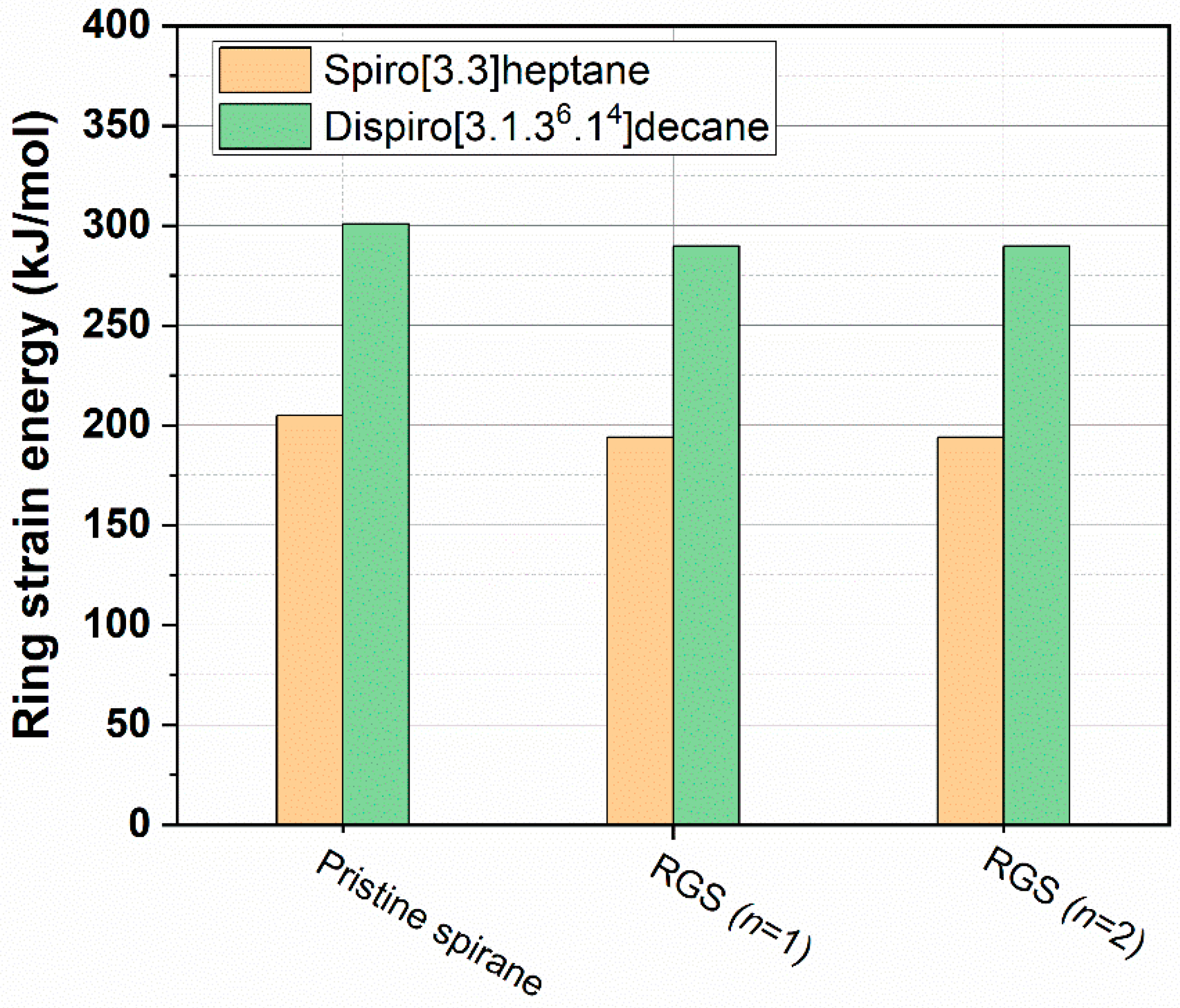
2.2. Prediction of Ring Strain Energy
2.3. Processing Performance of RGS
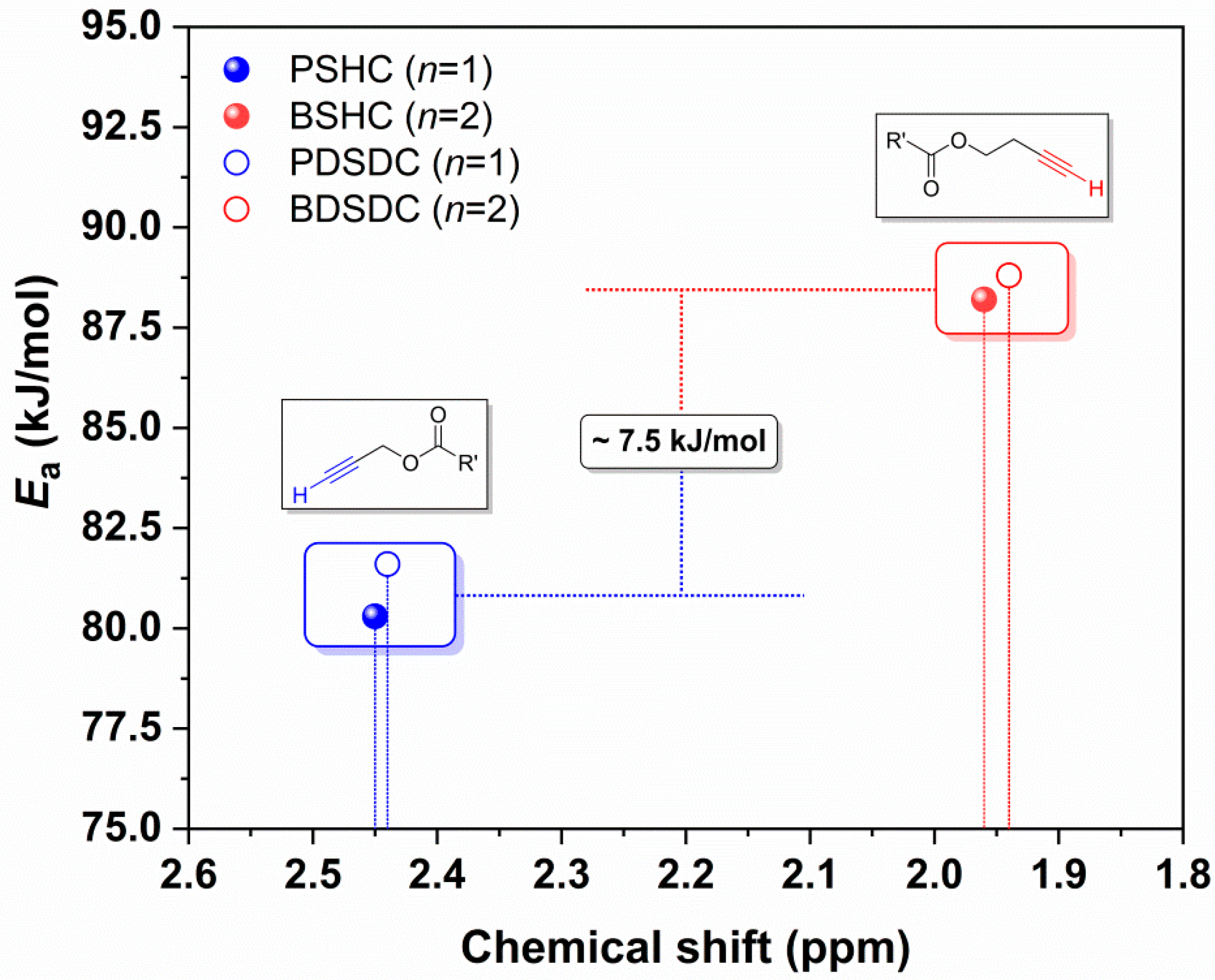
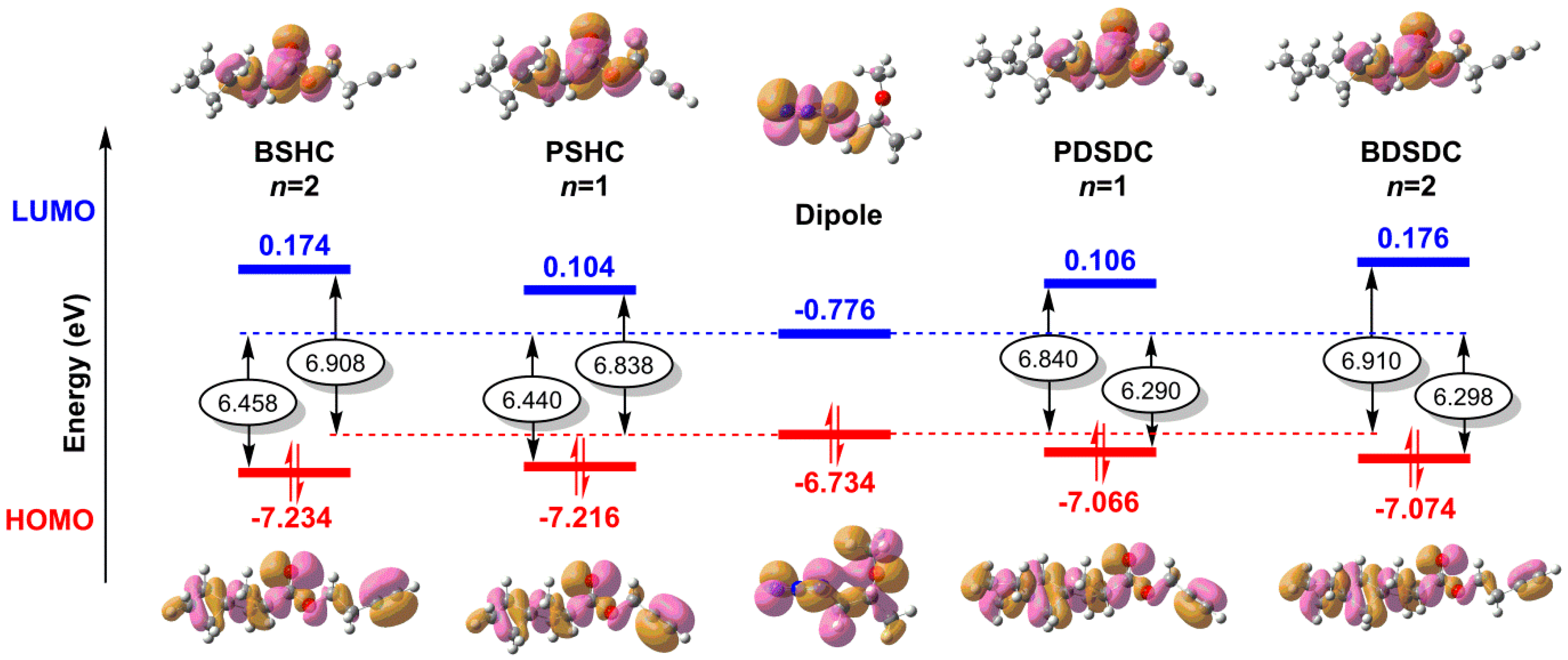
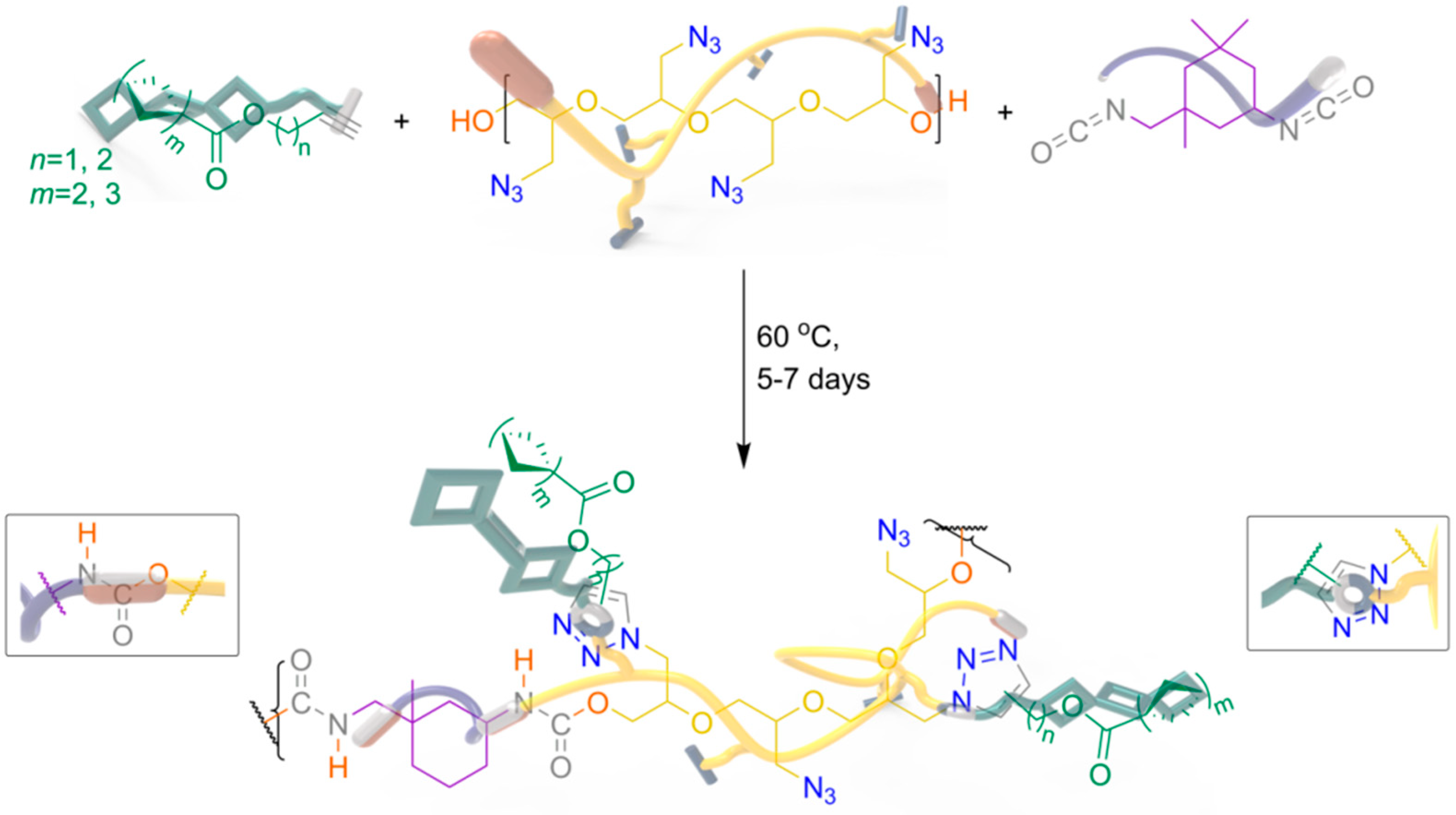
2.4. Catalyst-Free Click Reactivity
2.5. Thermal Stability
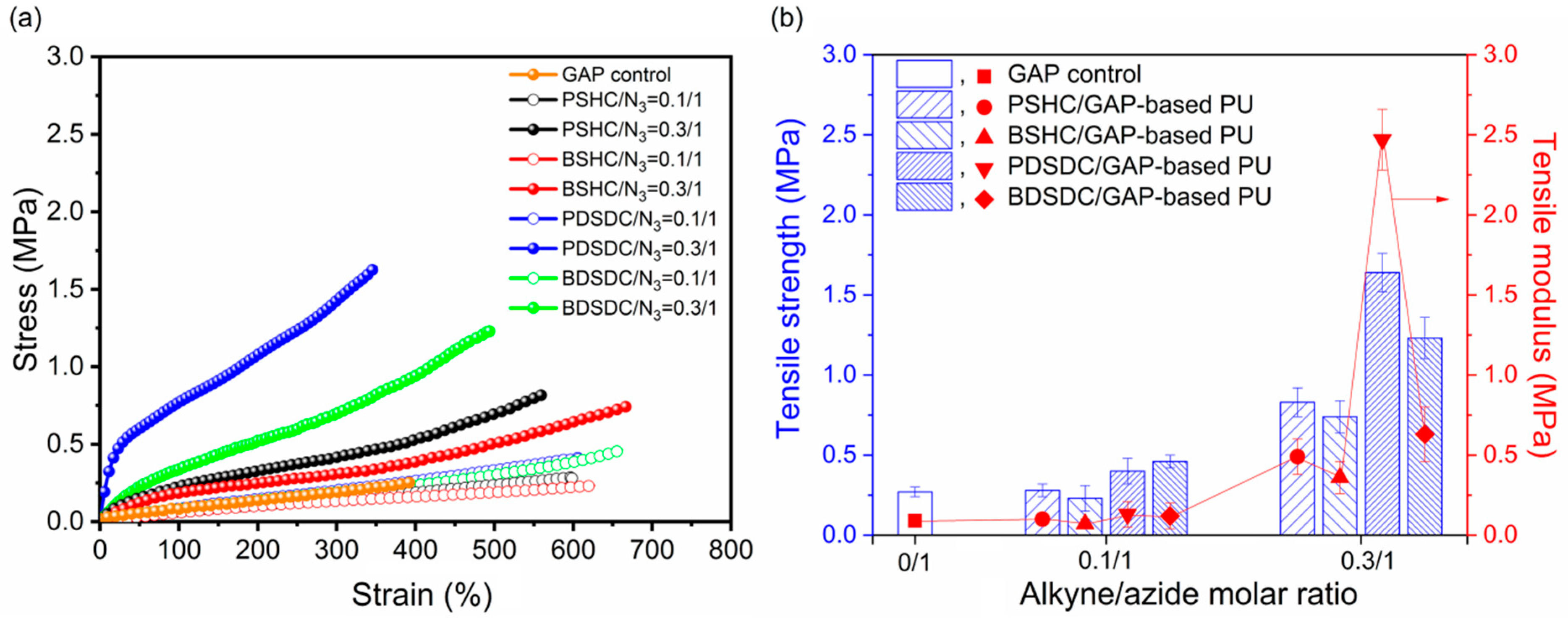
2.6. Tensile Properties
2.7. Thermochemical Properties
2.8. Impact Sensitivity
2.9. Migration Resistance
3. Experimental
3.1. Materials
3.2. Characterization
3.3. Synthesis of RGS
3.4. Fabrication of RGS@GAP-Based PUs
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Conflicts of Interest
References
- Bayer, O. Das Di-Isocyanat-Polyadditionsverfahren (Polyurethane). Angew. Chem. 1947, 59, 257–272. [Google Scholar] [CrossRef]
- Zukas, J.A.; Walters, W.P. Explosive Effects and Applications; Springer: New York, NY, USA, 1998. [Google Scholar]
- Yan, Q.L.; Zeman, S.; Elbeih, A. Recent advances in thermal analysis and stability evaluation of insensitive plastic bonded ex-plosives (PBXs). Thermochim. Acta 2012, 537, 1–12. [Google Scholar] [CrossRef]
- Bohn, M.A. Determination of the kinetic data of the thermal decomposition of energetic plasticizers and binders by adiabatic self heating. Thermochim. Acta 1999, 337, 121–139. [Google Scholar] [CrossRef]
- Kumari, D.; Balakshe, R.; Banerjee, S.; Singh, H. Energetic plasticizers for gun & rocket propellants. Rev. J. Chem. 2012, 2, 240–262. [Google Scholar] [CrossRef]
- Chen, Y.; Kwon, Y.; Kim, J.S. Synthesis and characterization of bis(2,2-dinitropropyl ethylene) formal plasticizer for energetic binders. J. Ind. Eng. Chem. 2012, 18, 1069–1075. [Google Scholar] [CrossRef]
- Provatas, A. Energetic Plasticizer Migration Studies. J. Energetic Mater. 2003, 21, 237–245. [Google Scholar] [CrossRef]
- Kwon, Y.H.; Kim, J.S.; Lee, B.J.; Bae, I.J. Ether-Based Reactive Plasticizer for Plastic Bonded Explosives. U.S. Patent 8,704,004, 30 January 2014. [Google Scholar]
- Kwon, Y.H.; Kim, J.S.; Lee, B.J.; Bae, I.J. Ester-Based Reactive Plasticizer for Plastic Bonded Explosives. U.S. Patent 8,816,124, 26 August 2014. [Google Scholar]
- Kwon, Y.H.; Kim, J.S.; Lee, B.J.; Bae, I.J. Ester-Based REACTIVE plasticizer for Plastic Bonded Explosives. U.S. Patent 8,940,922, 2 June 2015. [Google Scholar]
- Ma, M.; Shen, Y.; Kwon, Y.; Chung, C.; Kim, J.S. Reactive Energetic Plasticizers for Energetic Polyurethane Binders Prepared via Simultaneous Huisgen Azide-Alkyne Cycloaddition and Polyurethane Reaction. Propellants Explos. Pyrotech. 2016, 41, 746–756. [Google Scholar] [CrossRef]
- Boretos, J.W.; Pierce, W.S. Segmented Polyurethane: A New Elastomer for Biomedical Applications. Science 1967, 158, 1481–1482. [Google Scholar] [CrossRef]
- Fareghi-Alamdari, R.; Jafari, N.; Shahidzadeh, M.; Zekri, N. Reactive Plasticizers Covalently Linked to Glycidyl Azide Polymer via Catalyst-Free Huisgen Azide-Alkyne Cycloaddition. Propellants Explos. Pyrotech. 2018, 43, 893–897. [Google Scholar] [CrossRef]
- Ma, M.; Kwon, Y. Reactive Energetic Plasticizers Utilizing Cu-Free Azide-Alkyne 1,3-Dipolar Cycloaddition for In-Situ Prepa-ration of Poly(THF-co-GAP)-Based Polyurethane Energetic Binders. Polymers 2018, 10, 516. [Google Scholar] [CrossRef]
- Wang, B.; Qi, X.; Zhang, W.; Wang, K.; Li, W.; Zhang, Q. Synthesis of 1-(2H-tetrazol-5-yl)-5-nitraminotetrazole and its derivatives from 5-aminotetrazole and cyanogen azide: A promising strategy towards the development of C-N linked bistetrazolate en-ergetic materials. J. Mater. Chem. A 2017, 5, 20867–20873. [Google Scholar] [CrossRef]
- He, C.; Imler, G.H.; Parrish, D.A.; Shreeve, J.M. Energetic salts of 4-nitramino-3-(5-dinitromethyl-1,2,4-oxadiazolyl)-furazan: Powerful alliance towards good thermal stability and high performance. J. Mater. Chem. A 2018, 6, 16833–16837. [Google Scholar] [CrossRef]
- Sun, Q.; Li, X.; Lin, Q.; Lu, M. Dancing with 5-substituted monotetrazoles, oxygen-rich ions, and silver: Towards primary ex-plosives with positive oxygen balance and excellent energetic performance. J. Mater. Chem. A 2019, 7, 4611–4618. [Google Scholar] [CrossRef]
- Gao, H.; Shreeve, J.M. Azole-Based Energetic Salts. Chem. Rev. 2011, 111, 7377–7436. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Kim, J.S.; Kwon, Y. Synthesis and Properties of n-Al/Fluorinated Polyurethane Binders Prepared by Simultaneous Addition Polymerization and Catalyst-Free Azide-Alkyne Click Modification with Fluorinated Reactive Plasticizers. J. Nanosci. Nanotechnol. 2017, 17, 7344–7350. [Google Scholar] [CrossRef]
- Wiberg, K.B. The Concept of Strain in Organic Chemistry. Angew. Chem. Int. Ed. 1986, 25, 312–322. [Google Scholar] [CrossRef]
- Wright, M.E.; Allred, G.D.; Wardle, R.B.; Cannizzo, L.F. Polymers containing ring-strain energy. New monomers and polymers based on cyclopropane, norbornadiene, and quadricyclane. J. Org. Chem. 1993, 58, 4122–4126. [Google Scholar] [CrossRef]
- Eaton, P.E. Cubanes: Starting Materials for the Chemistry of the 1990s and the New Century. Angew. Chem. Int. Ed. 1992, 31, 1421–1436. [Google Scholar] [CrossRef]
- Eaton, P.E.; Zhang, M.X.; Gilardi, R.; Gelber, N.; Iyer, S.; Surapaneni, R. Octanitrocubane: A new nitrocarbon. Propellants Explos. Pyrotech. 2002, 27, 1–6. [Google Scholar] [CrossRef]
- Nair, U.R.; Sivabalan, R.; Gore, G.M.; Geetha, M.; Asthana, S.N.; Singh, H. Hexanitrohexaazaisowurtzitane (CL-20) and CL-20-based formulations (review). Combust. Explos. Shock. Waves 2005, 41, 121–132. [Google Scholar] [CrossRef]
- Kao, J.; Radom, L. An ab initio molecular orbital study of structures and energies of spirocompounds: Spiro [3.3] heptane and spiro [3.3] hepta-1, 5-diene. Tetrahedron 1978, 34, 2515–2521. [Google Scholar] [CrossRef]
- Hoffmann, R.; Imamura, A.; Zeiss, G.D. Spirarenes. J. Am. Chem. Soc. 1967, 89, 5215–5220. [Google Scholar] [CrossRef]
- Wynberg, H.; Houbiers, J.P.M. Attempted assignment of absolute configuration to the d-Fecht acid and other 2,6-disubstituted spiro[3.3]heptane derivatives. J. Org. Chem. 1971, 36, 834–842. [Google Scholar] [CrossRef]
- Chan, L.K.M.; Gemmell, P.A.; Gray, G.W.; Lacey, D.; Toyne, K.J. Synthesis and liquid crystal properties of compounds in-corporating cyclobutane, spiro [3.3] heptane and dispiro [3.1. 3.1] decane rings. Mol. Cryst. Liq. Cryst. 1987, 147, 113–139. [Google Scholar] [CrossRef]
- D’Yakonov, V.A.; Finkelshtein, E.S.; Ibragimov, A.G. Dzhemilev reaction for the synthesis of spiro[3.3]heptane and spiro[3.4]octanes. Tetrahedron Lett. 2007, 48, 8583–8586. [Google Scholar] [CrossRef]
- Radchenko, D.S.; Grygorenko, O.O.; Komarov, I.V. Synthesis of conformationally restricted glutamic acid analogs based on the spiro[3.3]heptane scaffold. Tetrahedron Asymmetry 2008, 19, 2924–2930. [Google Scholar] [CrossRef]
- Reiffenrath, V.; Bremer, M.; Klasen-Memmer, M. Cyclobutane and Spiro [3.3] Heptane Compounds. U.S. Patent 7,744,968, 30 September 2009. [Google Scholar]
- Burkhard, J.A.; Guérot, C.; Knust, H.; Carreira, E.M. Expanding the Azaspiro[3.3]heptane Family: Synthesis of Novel Highly Functionalized Building Blocks. Org. Lett. 2011, 14, 66–69. [Google Scholar] [CrossRef]
- Koch, S.D. Dispirane Hydrocarbons as High Energy Fuels. U.S. Patent 3,113,421, 10 December 1963. [Google Scholar]
- Wineman, R.J. Dispirane Hydrocarbons as New Compounds and Use as High Energy Fuels. U.S. Patent 3,113,422, 10 December 1963. [Google Scholar]
- Buchta, E.; Geibel, K. Spirocyclische Verbindungen, I. Spirane, Di-, Tri- und Tetraspirane. Eur. J. Org. Chem. 1961, 648, 36–50. [Google Scholar] [CrossRef]
- Khoury, P.R.; Goddard, J.D.; Tam, W. Ring strain energies: Substituted rings, norbornanes, norbornenes and norbornadienes. Tetrahedron 2004, 60, 8103–8112. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 09, Revision A. 02; Gaussian, Inc.: Wallingford, CT, USA, 2009. [Google Scholar]
- Ma, M.; Kwon, Y. Preparation of energetic polyurethane binders with enhanced properties by nonmigratory reactive monocyclic plasticizers. Eur. Polym. J. 2019, 123, 109414. [Google Scholar] [CrossRef]
- Ma, M.; Shen, Y.; Kwon, Y. Fabrication of polyurethane binders grafted with functional reactive plasticizers by a catalyst-free click reaction. J. Polym. Sci. 2020, 58, 402–411. [Google Scholar] [CrossRef]
- Bach, R.D.; Dmitrenko, O. The Effect of Substitutents on the Strain Energies of Small Ring Compounds. J. Org. Chem. 2002, 67, 2588–2599. [Google Scholar] [CrossRef] [PubMed]
- Wingborg, N.; Eldsäter, C. 2,2-Dinitro-1,3-Bis-Nitrooxy-Propane (NPN): A New Energetic Plasticizer. Propellants Explos. Pyrotech. 2002, 27, 314–319. [Google Scholar] [CrossRef]
- Ward, M.I.; Sweeney, J. An Introduction to the Mechanical Properties of Solid Polymers; John Wiley & Sons Ltd.: Chichester, UK, 2004. [Google Scholar]
- Wypych, G. Handbook of Plasticizers; ChemTec Publishing: Toronto, ON, Canada, 2004. [Google Scholar]
- Min, B.S.; Jeon, H.B.; Jeong, T.U.; Kim, S.Y. Energetic polymeric networks prepared via a solvent- and catalyst-free thermal cycloaddition of azide-bearing polymers with alkynes and hydroxyl-isocyanate addition reactions. Polym. Chem. 2015, 6, 7913–7920. [Google Scholar] [CrossRef]
- ANSI/ASTM E698-79; Standard Test Method for Arrhenius Kinetic Constant for Thermally Unstable Materials. ASTM: Philadelphia, PA, USA, 1979.
- Doyle, C.D. Estimating isothermal life from thermogravimetric data. J. Appl. Polym. Sci. 1962, 6, 639–642. [Google Scholar] [CrossRef]
- Gorman, I.E.; Willer, R.L.; Kemp, L.K.; Storey, R.F. Development of a triazole-cure resin system for composites: Evaluation of alkyne curatives. Polymer 2012, 53, 2548–2558. [Google Scholar] [CrossRef]
- Chambers, R.D. Fluorine in Organic Chemistry; Blackwell Publishing Ltd.: Oxford, UK, 2004. [Google Scholar]
- Ma, M.; Kwon, Y. Reactive cycloalkane plasticizers covalently linked to energetic polyurethane binders via facile control of an in situ Cu-free azide–alkyne 1,3-dipolar cycloaddition reaction. Polym. Chem. 2018, 9, 5452–5461. [Google Scholar] [CrossRef]
- Landsem, E.; Jensen, T.L.; Kristensen, T.E.; Hansen, F.K.; Benneche, T.; Unneberg, E. Isocyanate-Free and Dual Curing of Smokeless Composite Rocket Propellants. Propellants Explos. Pyrotech. 2012, 38, 75–86. [Google Scholar] [CrossRef]
- Gaur, B.; Lochab, B.; Choudhary, V.; Varma, I.K. Azido Polymers—Energetic Binders for Solid Rocket Propellants. J. Macromol. Sci. Part C Polym. Rev. 2003, 43, 505–545. [Google Scholar] [CrossRef]
- Selim, K.; Yilmaz, L. Thermal characterization of glycidyl azide polymer (GAP) and GAP-based binders for composite propellants. J. Appl. Polym. Sci. 2000, 77, 538–546. [Google Scholar] [CrossRef]
- Min, B.S. Characterization of the Plasticized GAP/PEG and GAP/PCL Block Copolyurethane Binder Matrices and its Propel-lants, Propellants. Explos. Pyrotech. 2008, 2, 131–138. [Google Scholar] [CrossRef]
- Diaz, E.; Brousseau, P.; Ampleman, G.; Prud’Homme, R.E. Heats of Combustion and Formation of New Energetic Thermoplastic Elastomers Based on GAP, PolyNIMMO and PolyGLYN. Propellants Explos. Pyrotech. 2003, 28, 101–106. [Google Scholar] [CrossRef]
- Gibbs, T.R.; Popolato, A. LASL Explosive Property Data; University of California Press: Berkeley, CA, USA, 1980. [Google Scholar]
- Klapötke, T.M. Chemistry of High-Energy Materials; Walter de Gruyter GmbH: Berlin, Germany; Boston, MA, USA, 2015. [Google Scholar]
- Agrawal, J.P. High Energy Materials: Propellants, Explosives and Pyrotechnics; John Wiley & Sons: Hoboken, NJ, USA, 2010. [Google Scholar]
- Eroǧlu, M.S.; Baysal, B.M.; Güven, O. Determination of solubility parameters of poly(epichlorohydrin) and poly(glycidyl azide) networks. Polymer 1997, 38, 1945–1947. [Google Scholar] [CrossRef]









Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ma, M.; Kwon, Y. Simultaneous Enhancement of the Mechanical Properties, Performance and Insensitivity of an Energetic Elastomeric Polyurethane Binder by Kinetically Grafting Reactive Spiranes. Polymers 2023, 15, 4564. https://doi.org/10.3390/polym15234564
Ma M, Kwon Y. Simultaneous Enhancement of the Mechanical Properties, Performance and Insensitivity of an Energetic Elastomeric Polyurethane Binder by Kinetically Grafting Reactive Spiranes. Polymers. 2023; 15(23):4564. https://doi.org/10.3390/polym15234564
Chicago/Turabian StyleMa, Mingyang, and Younghwan Kwon. 2023. "Simultaneous Enhancement of the Mechanical Properties, Performance and Insensitivity of an Energetic Elastomeric Polyurethane Binder by Kinetically Grafting Reactive Spiranes" Polymers 15, no. 23: 4564. https://doi.org/10.3390/polym15234564