

Pyrolysis of Automotive Shredder Residue (ASR): Thermogravimetry, In-Situ Synchrotron IR and Gas-Phase IR of Polymeric Components
 , and
, and
Abstract
:1. Introduction
- (a)
- Morphology and chemical characteristics of ASR and its components.
- (b)
- Thermal analyses of ASR components for understanding the degradation kinetics and identification of the critical temperatures or temperature ranges of ASR pyrolysis. This includes synchrotron-based IR spectroscopy to examine the evolution of surface functional groups and thermogravimetric-IR to identify the gases evolved at different temperatures and heating rates. The resulting data were compared with those in the literature to characterise the active functional groups and evolved gases. For the first time, this study provides the information of surface functional groups evolved in situ during ASR pyrolysis using synchrotron IR.
- (c)
- To obtain the kinetic parameters using multistage model.
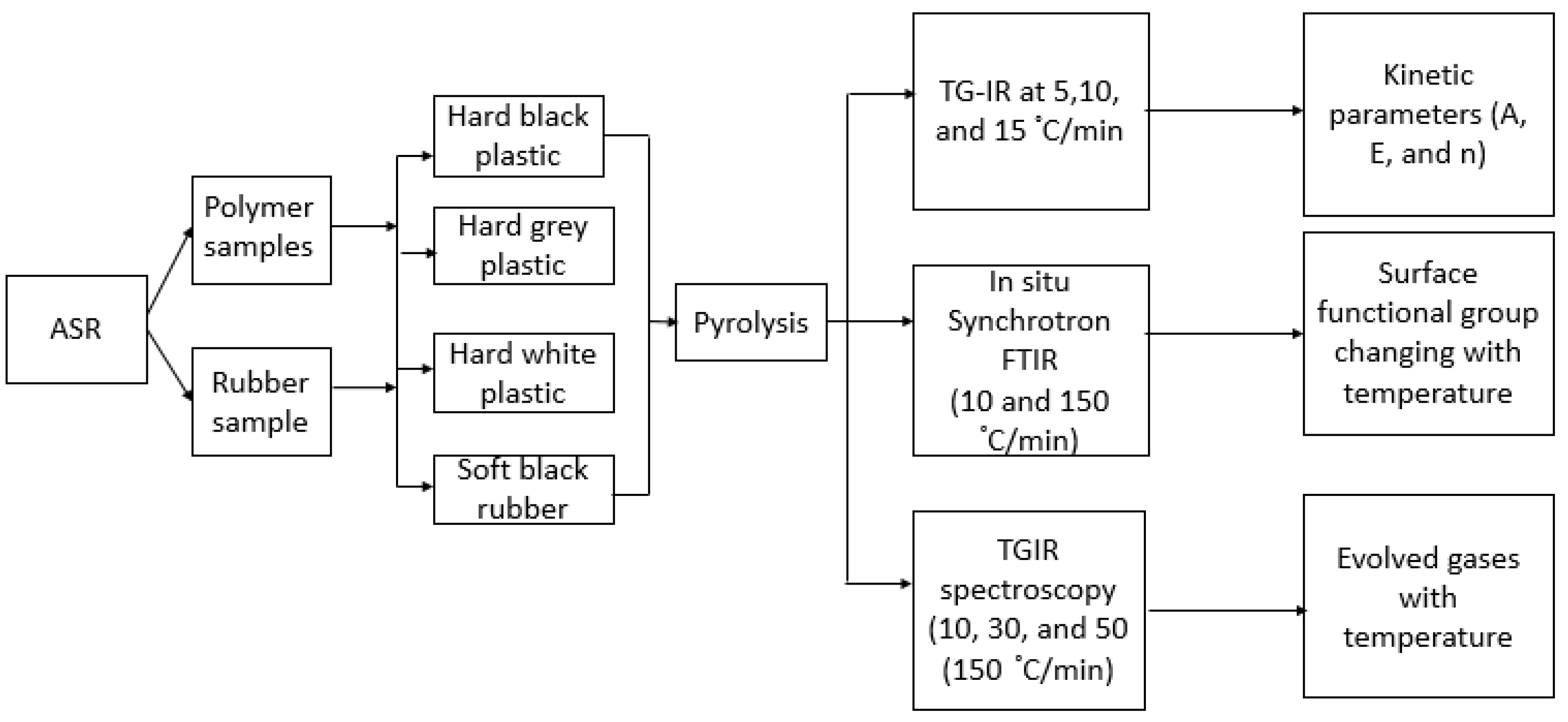
2. Materials and Methods
2.1. Introduction
2.2. Characterization of Feedstock
2.2.1. Scanning Electron Microscopy (SEM) and Energy Dispersive X-ray Spectroscopy (EDX)
2.2.2. Thermogravimetric Analysis (TGA) and Elemental Analysis
2.2.3. Simultaneous Thermogravimetric and Gas-phase Infrared Spectroscopy (TG-IR)
2.2.4. In Situ Synchrotron FT-IR Spectroscopy
2.2.5. X-ray Fluorescence (XRF) Analysis
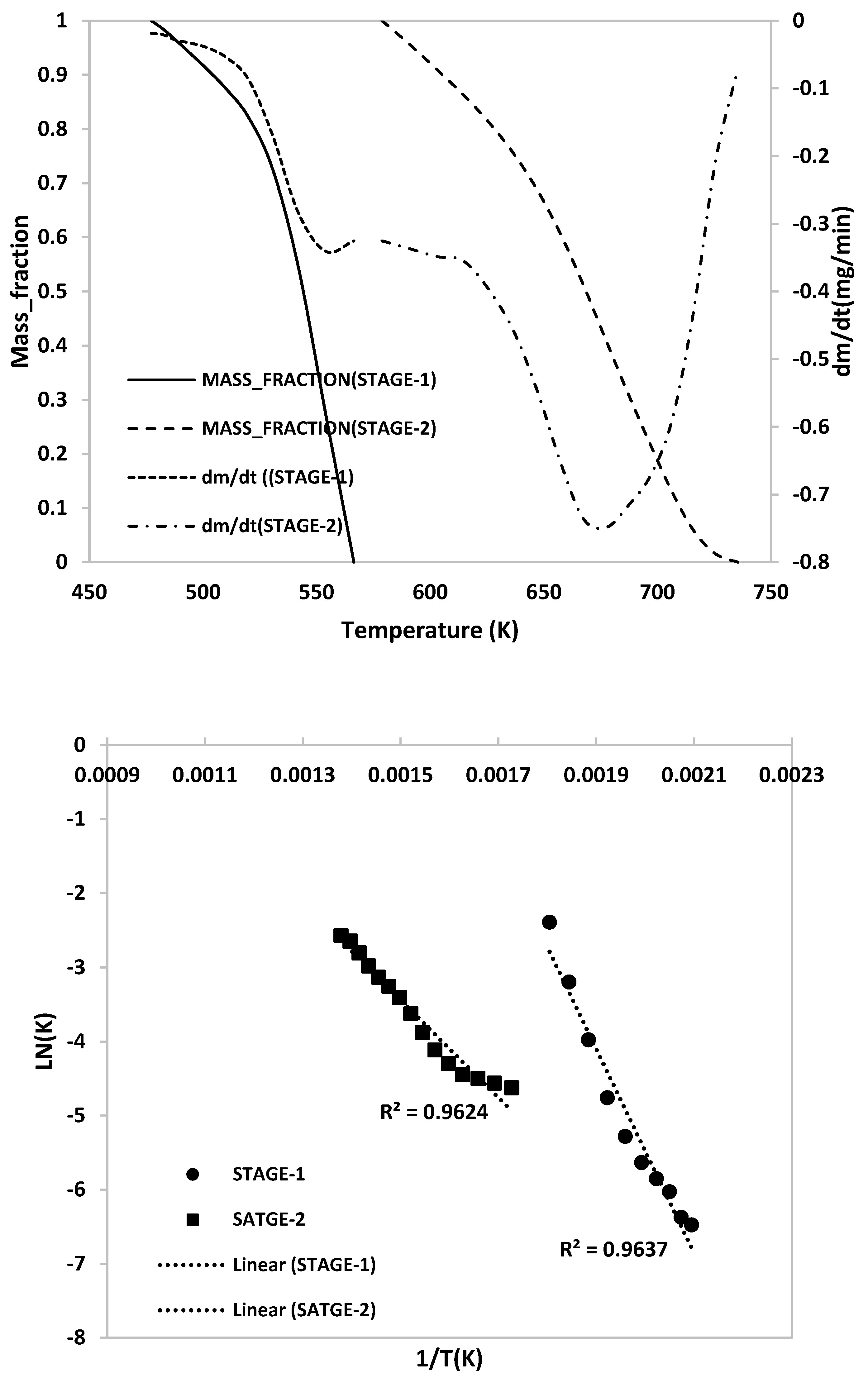
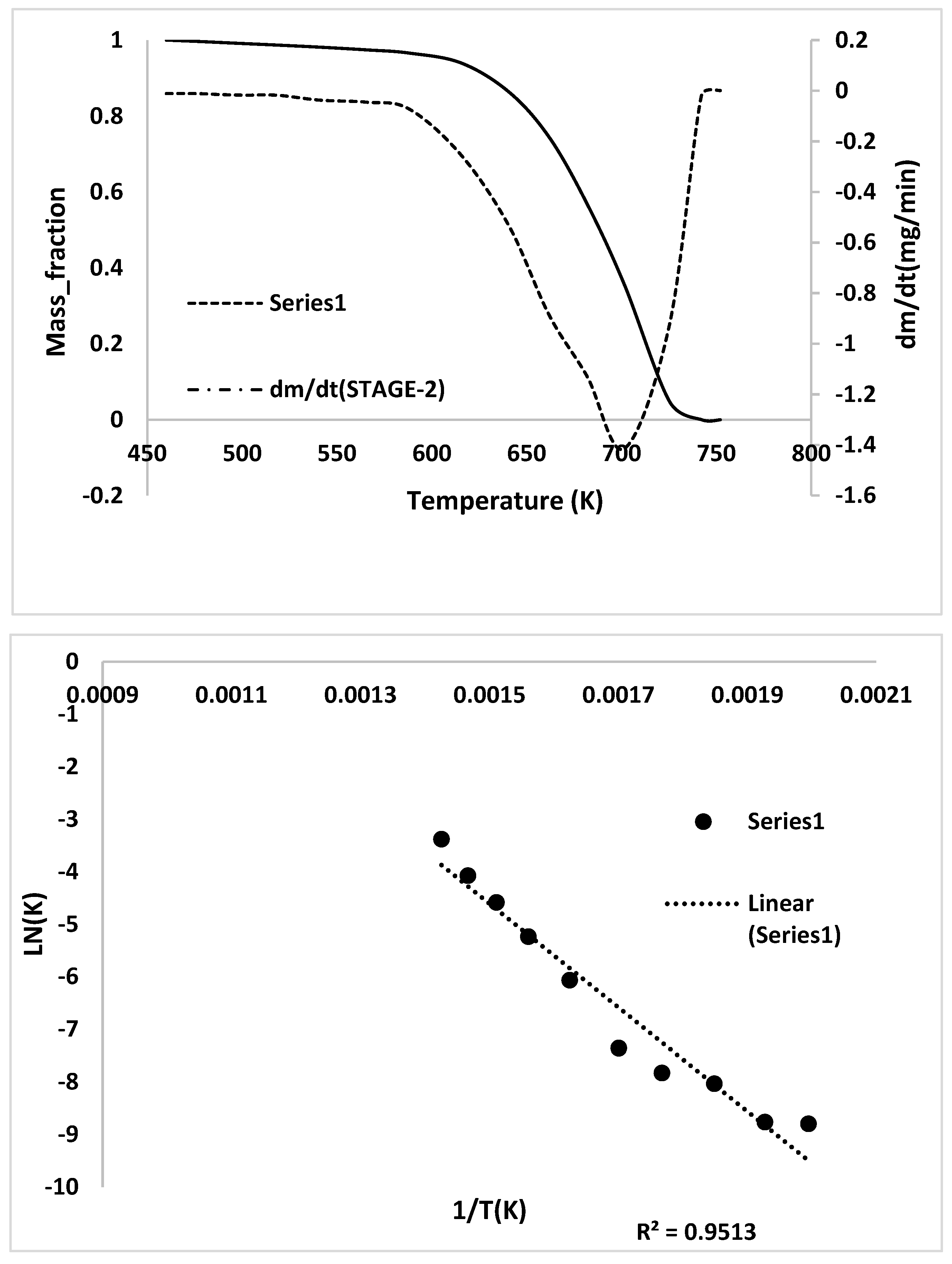
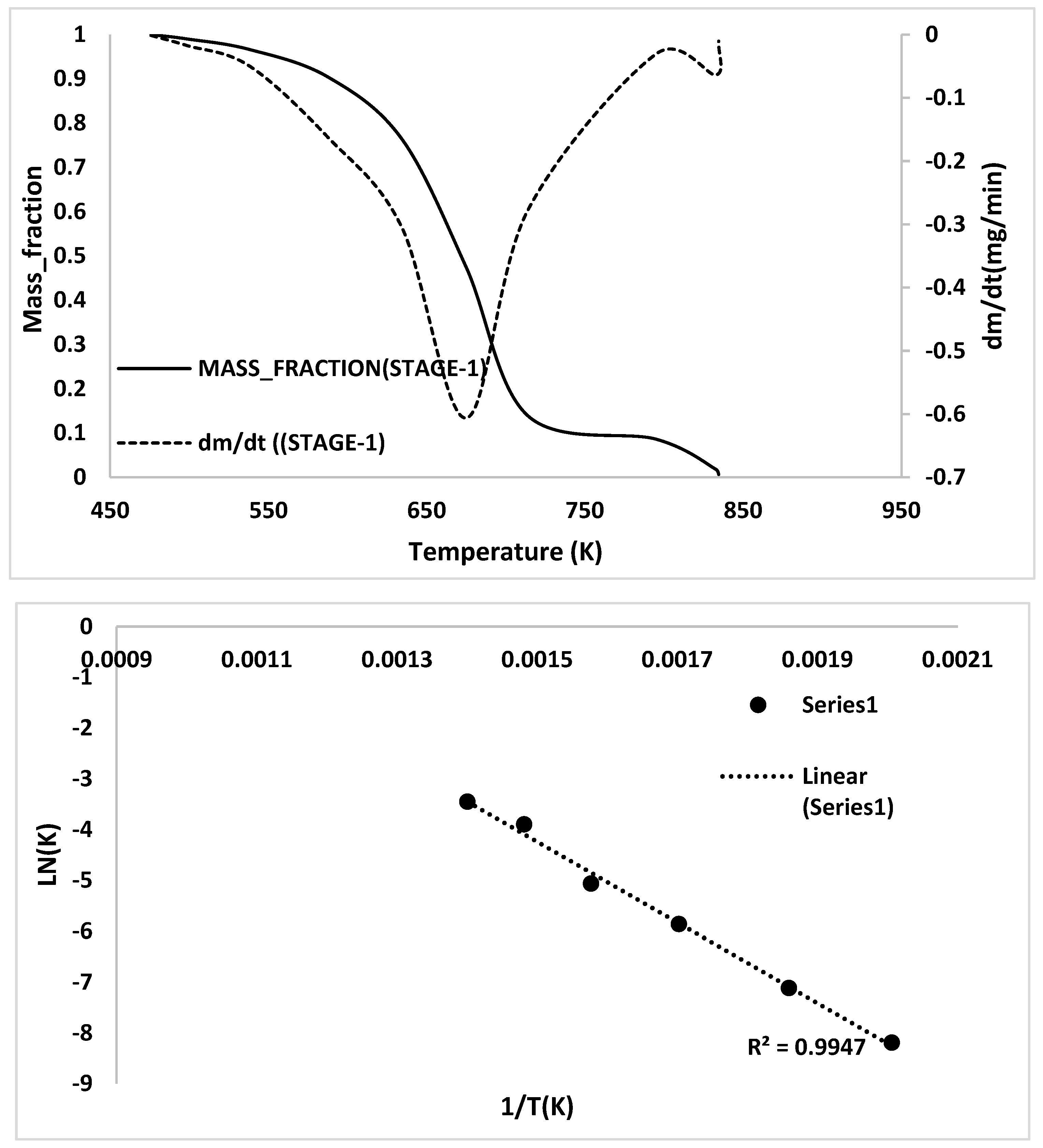
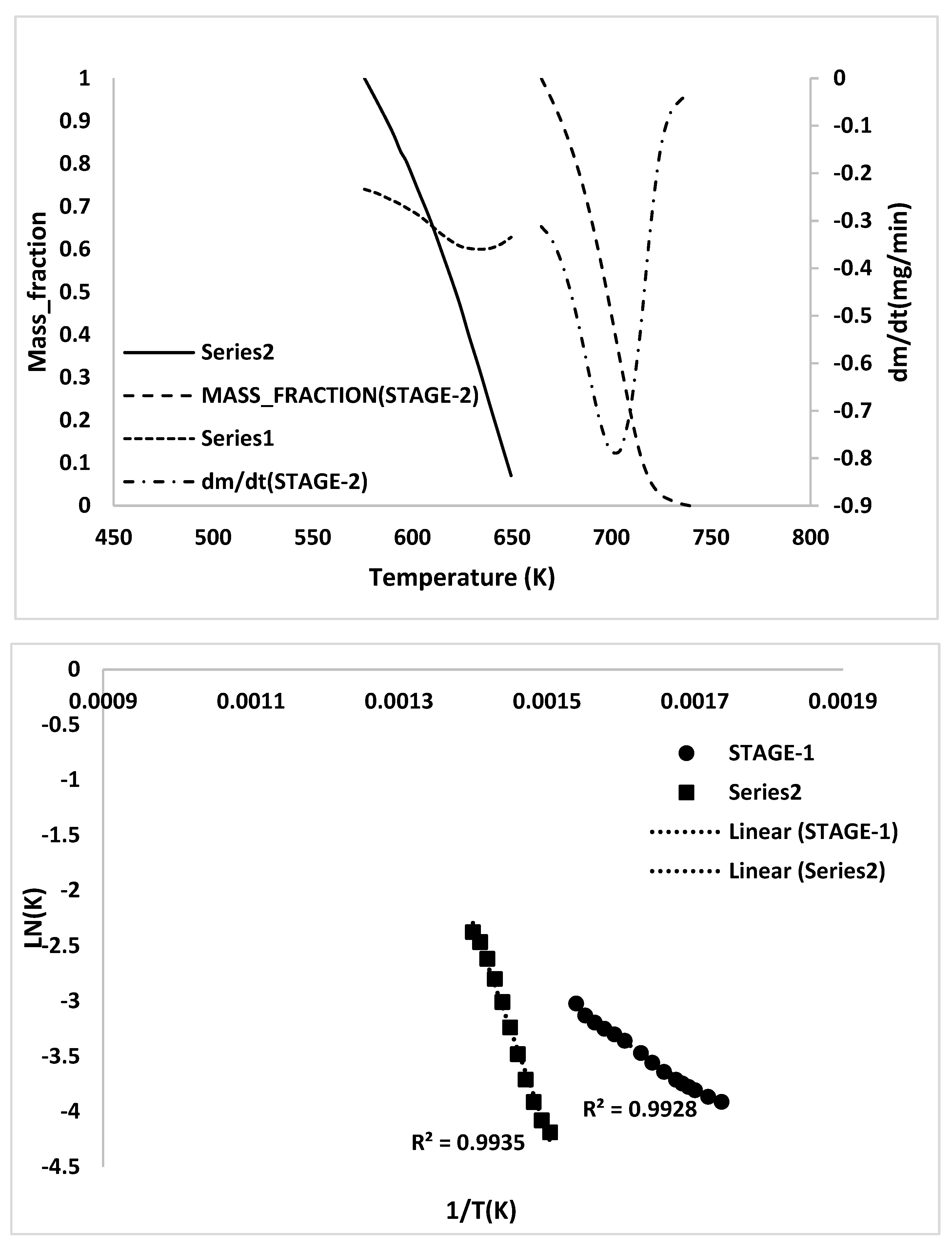
2.3. Kinetic Analysis
3. Results and Discussions
3.1. Proximate and Ultimate Analysis
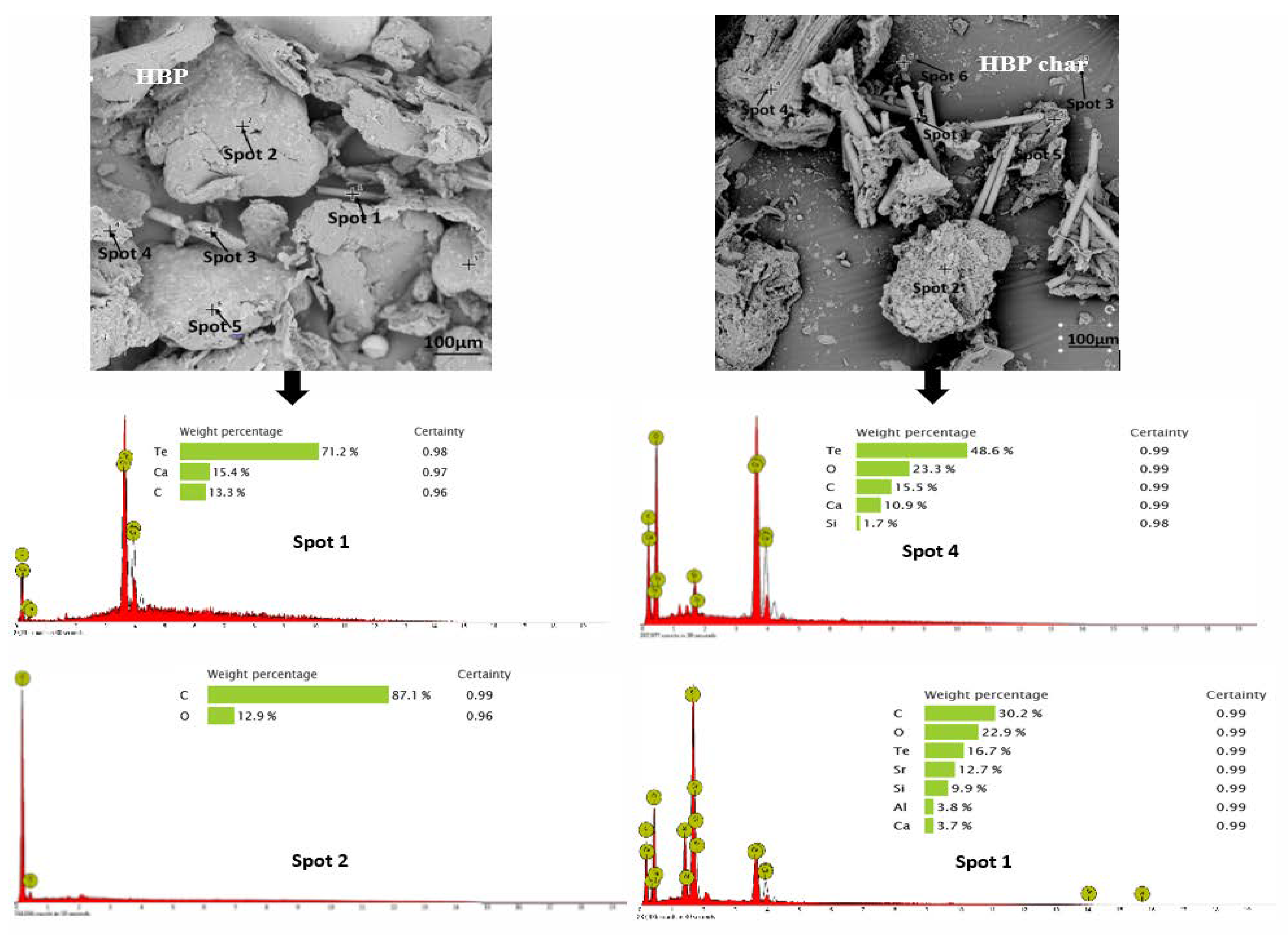
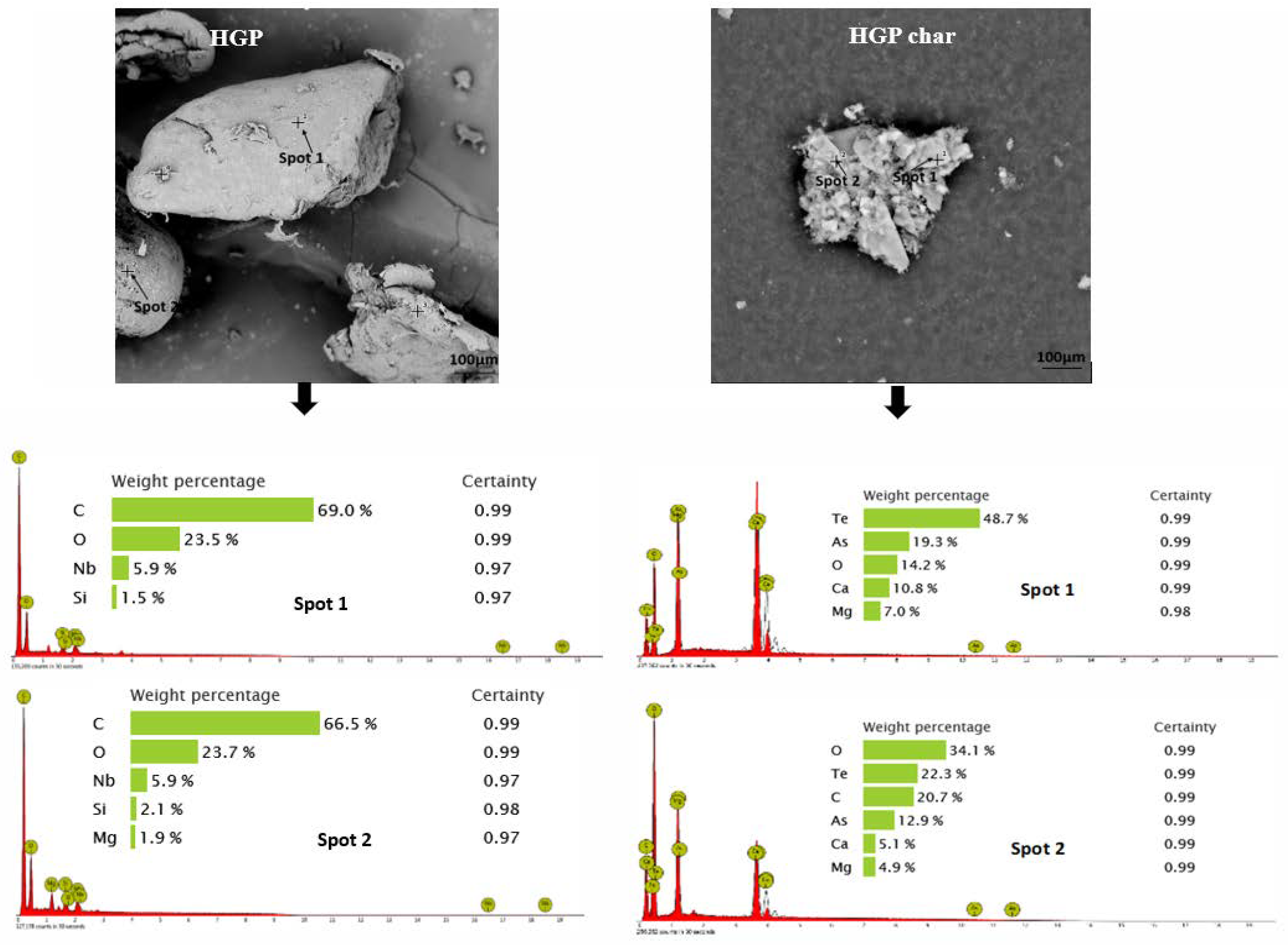
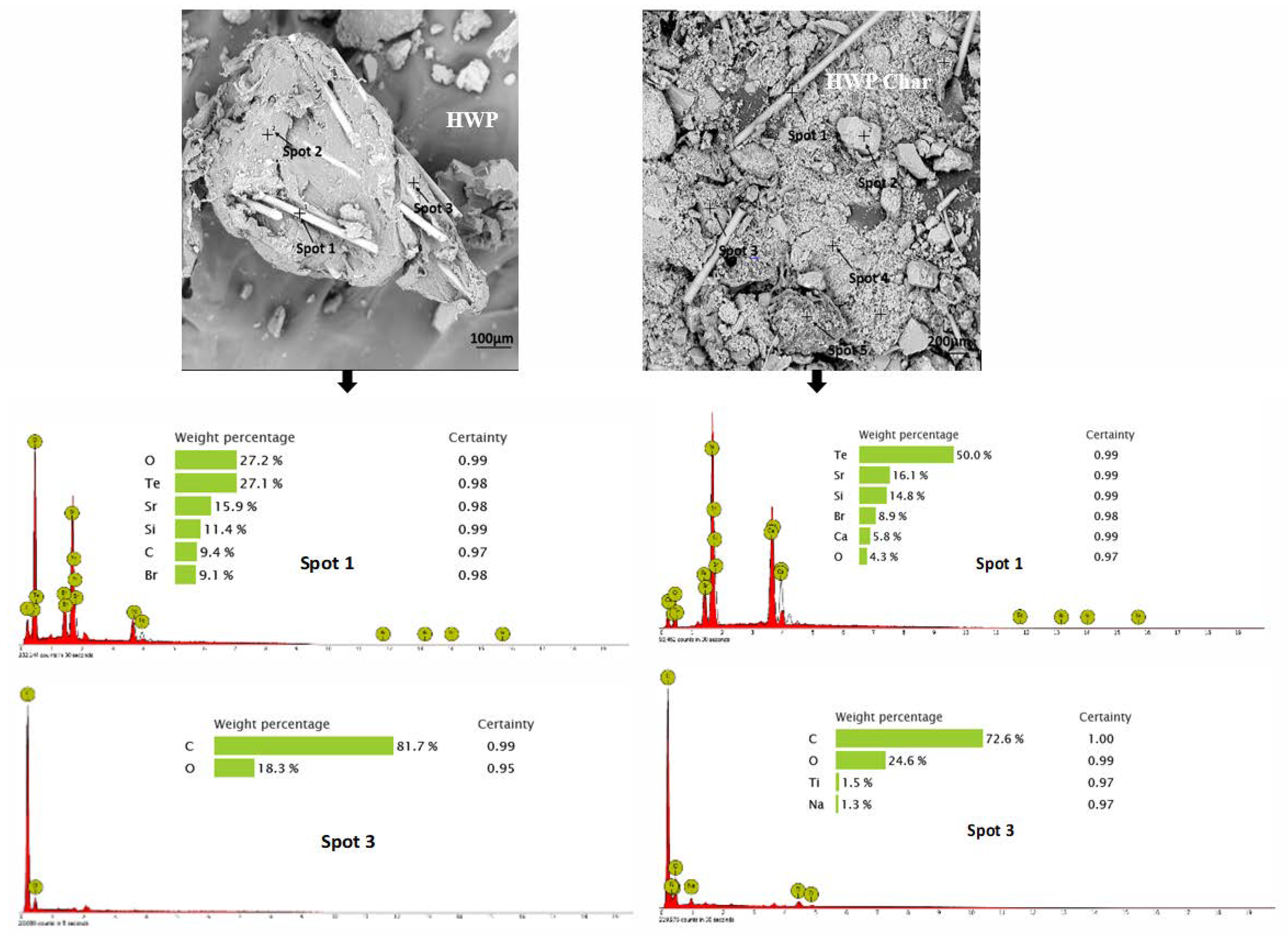
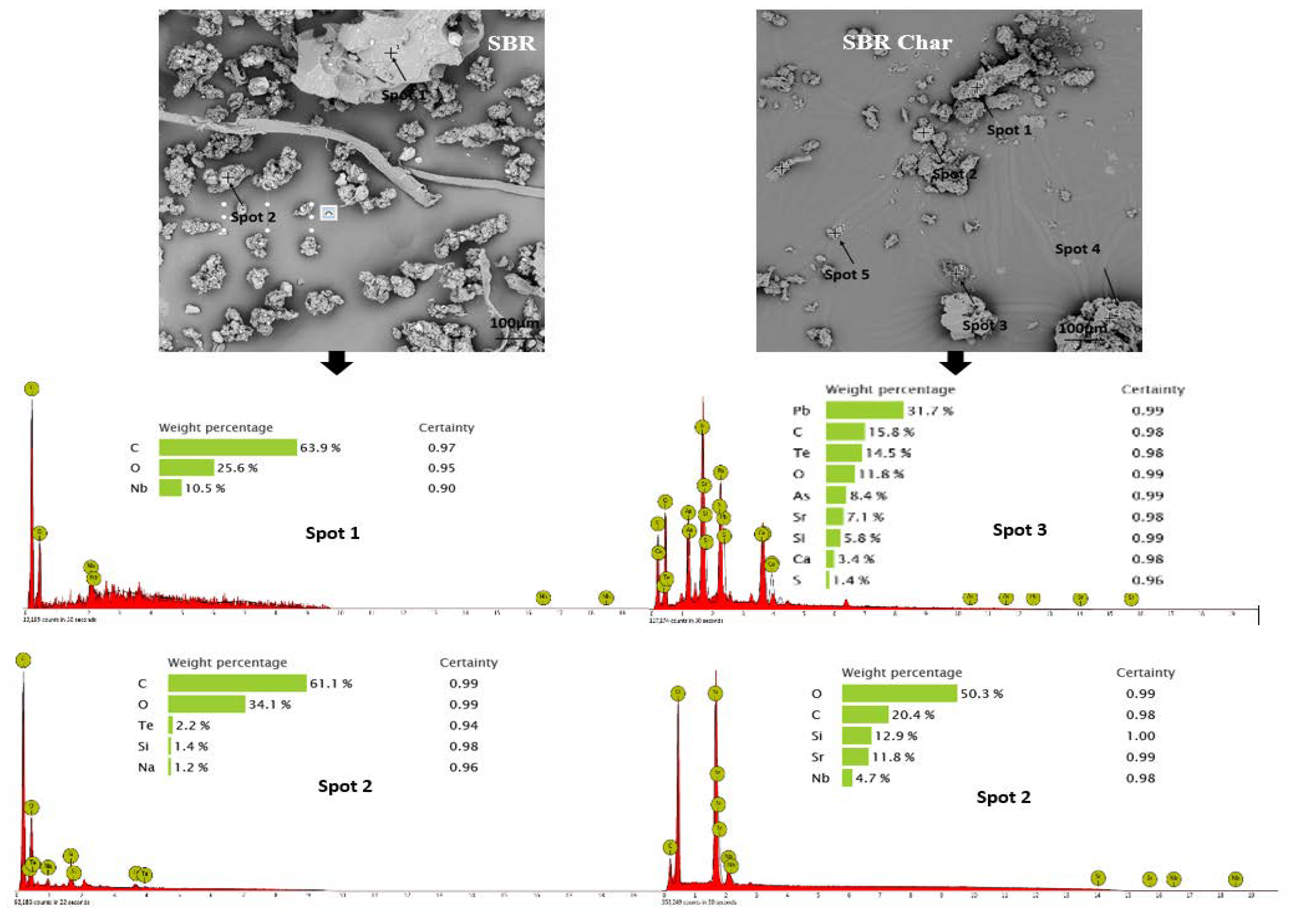
3.2. Morphology and Broad Chemical Composition of Different ASR Components and Their Pyrolysis Products
3.3. TG-IR and In Situ Synchrotron FTIR of Different ASR Components
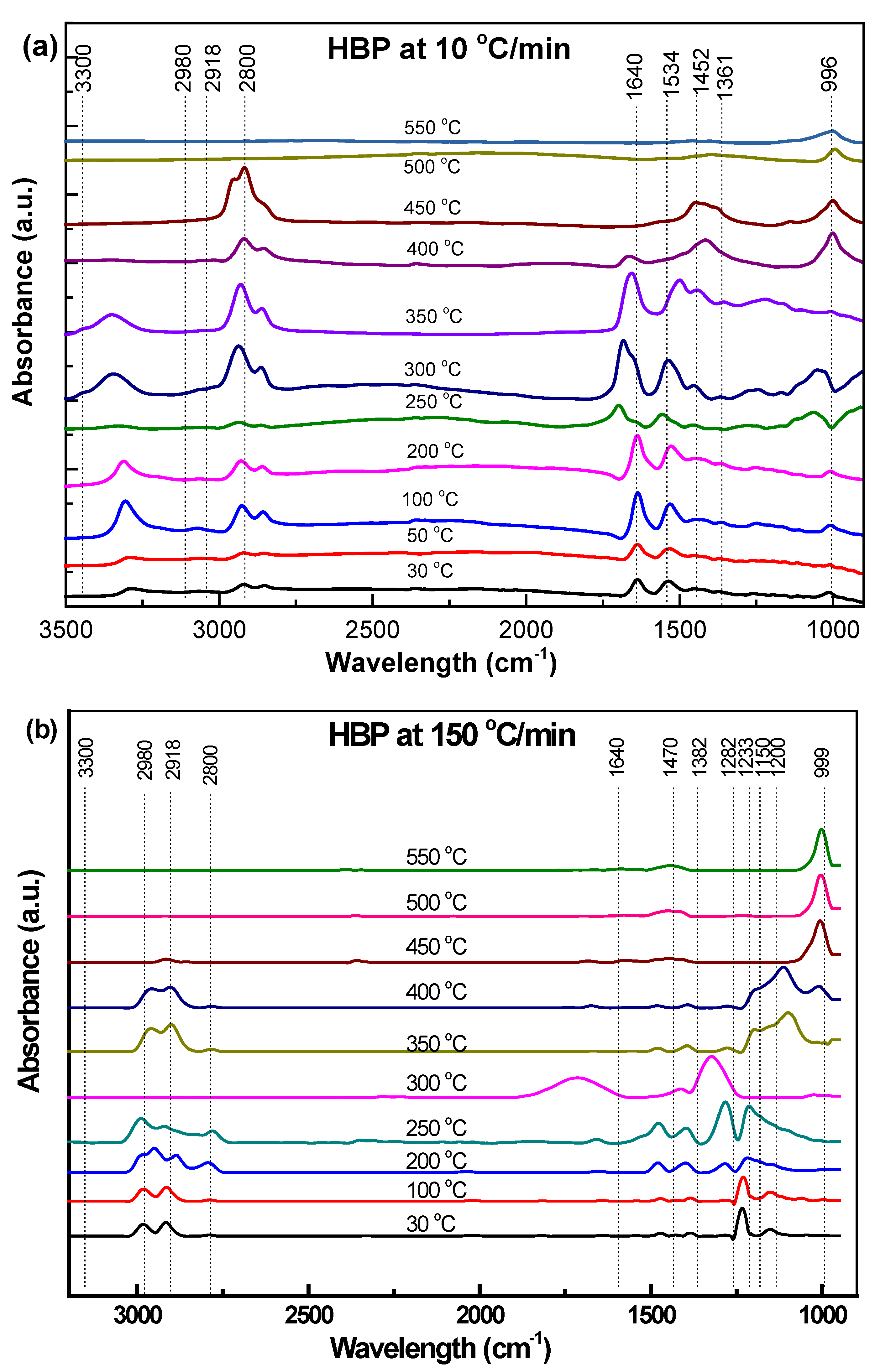
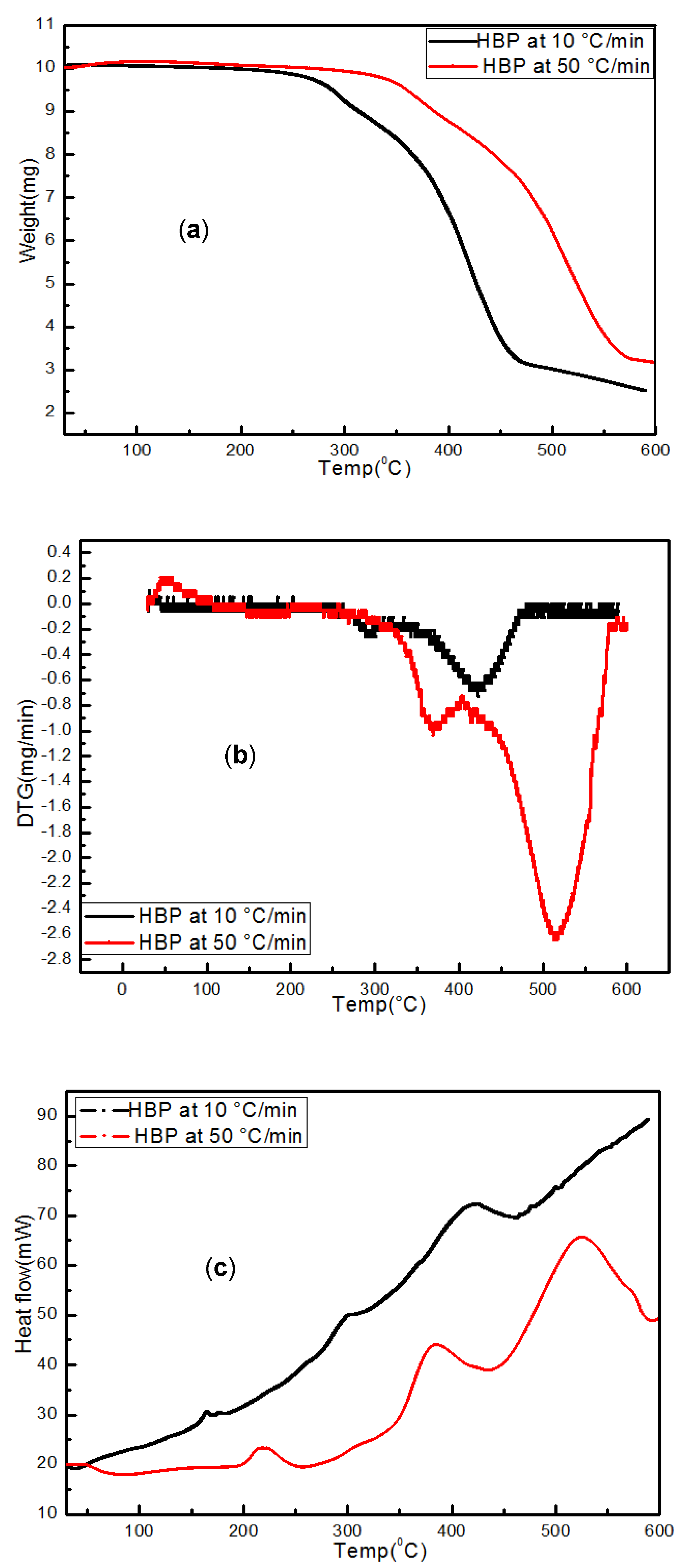
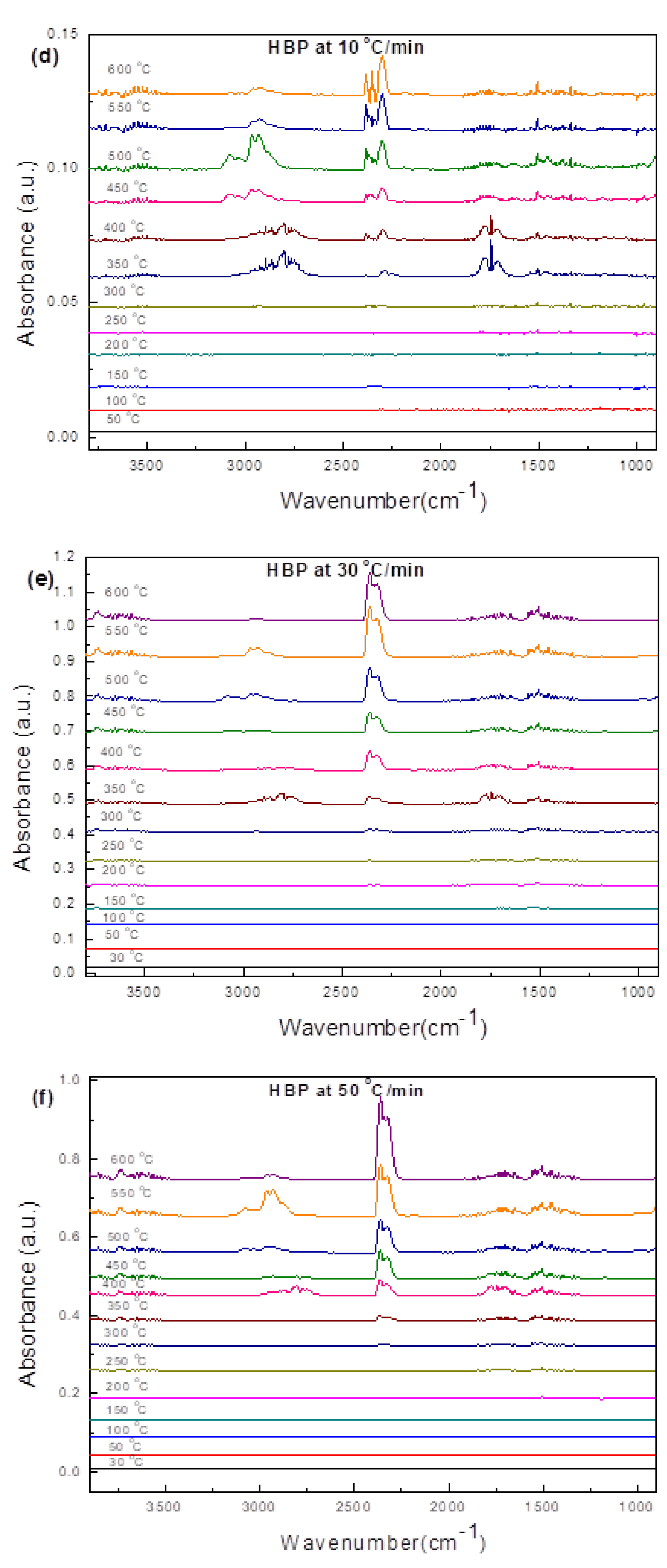
3.3.1. Hard Black Plastic (HBP)
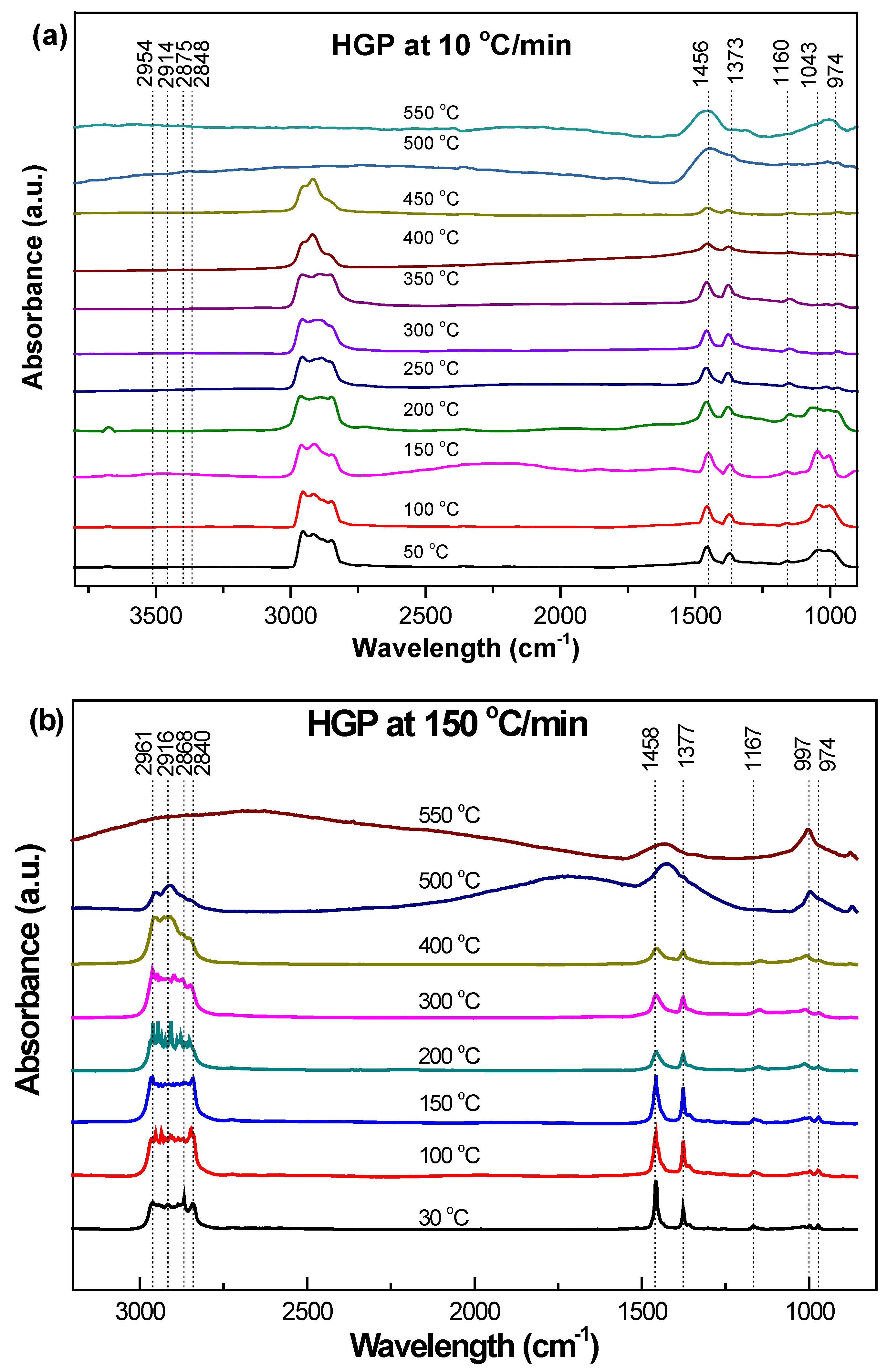
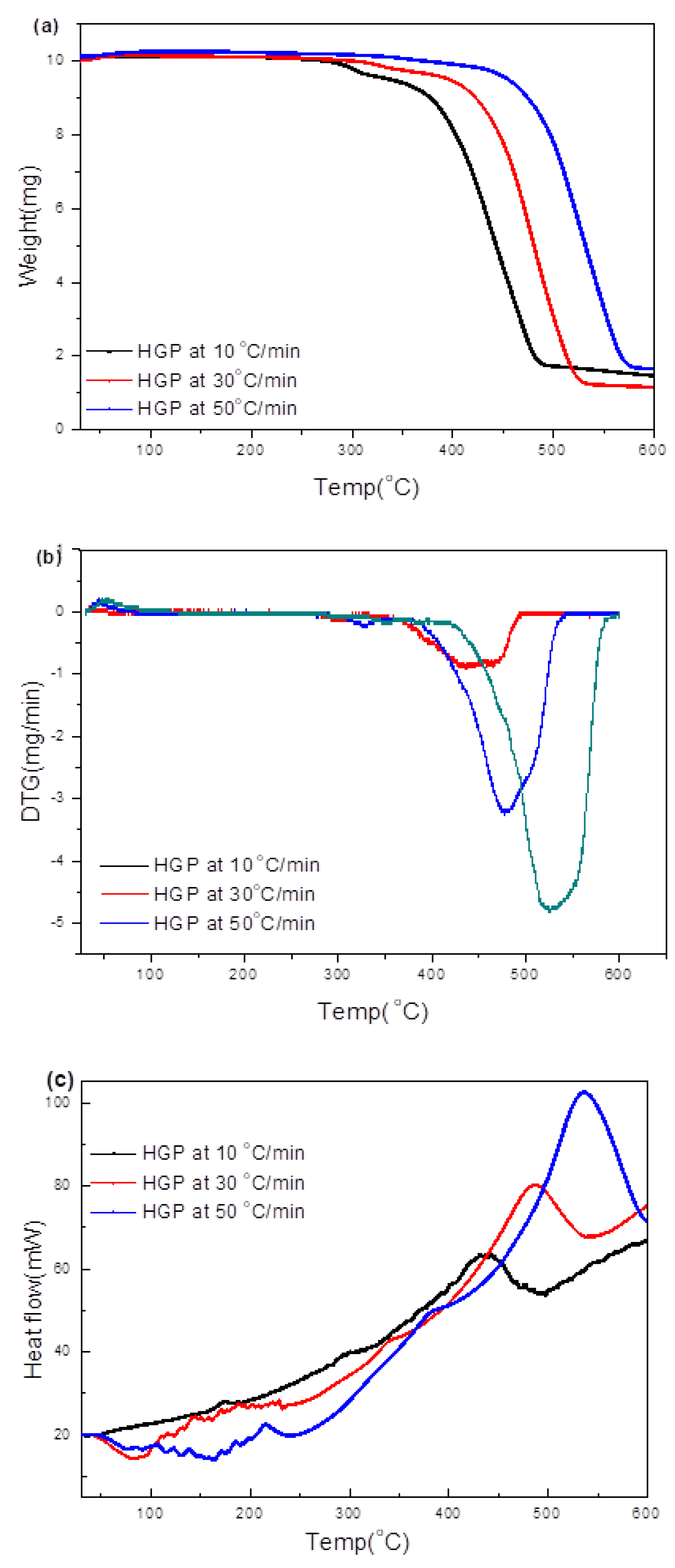
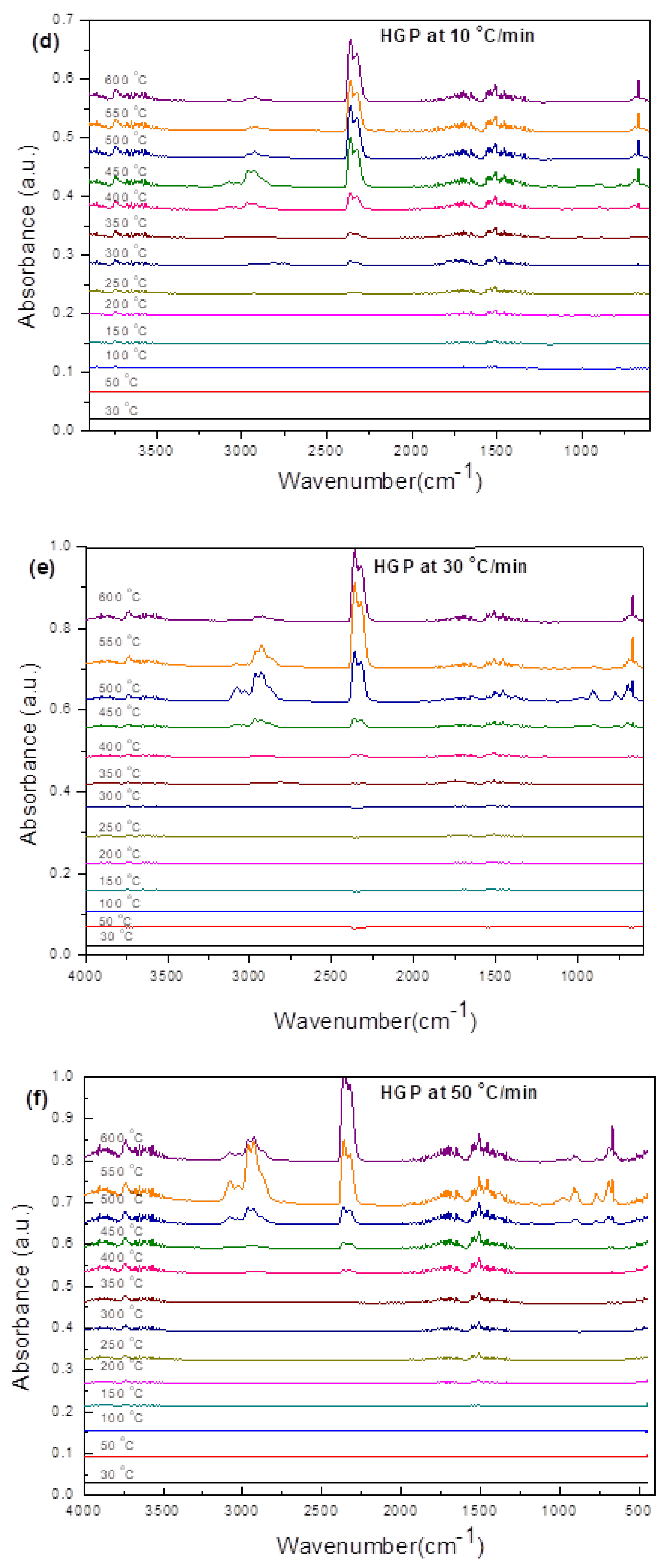
3.3.2. Hard Grey Plastic (HGP)
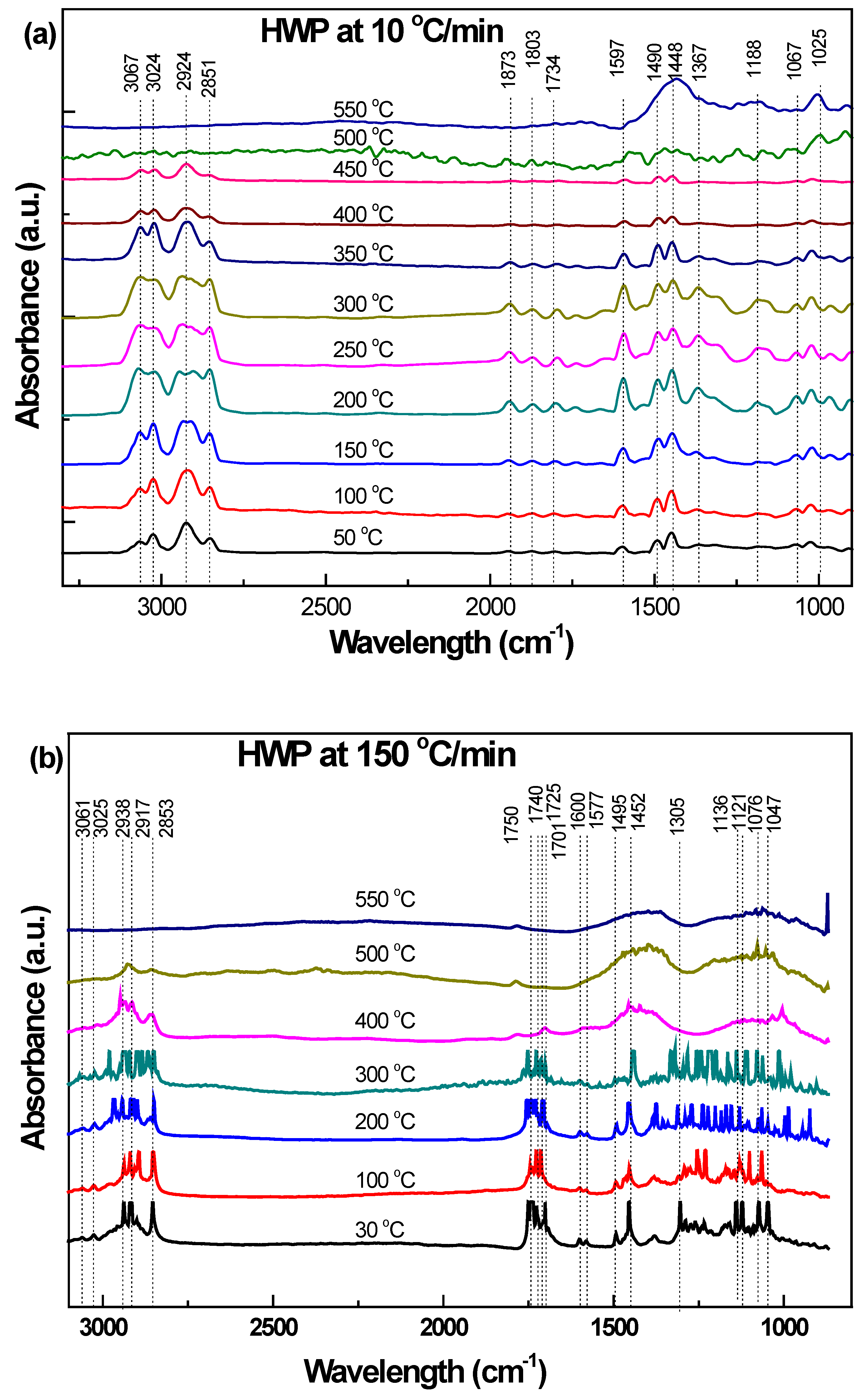
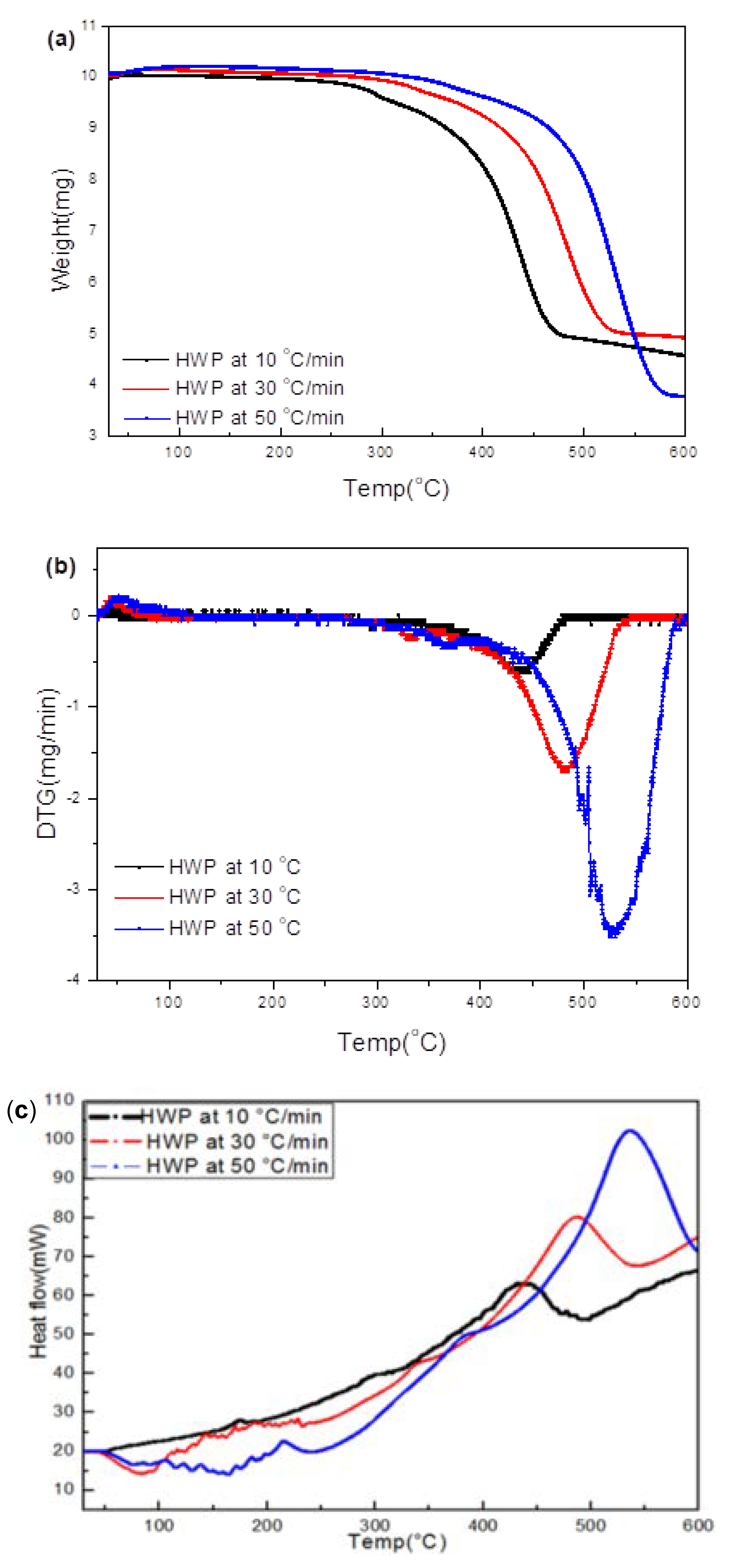
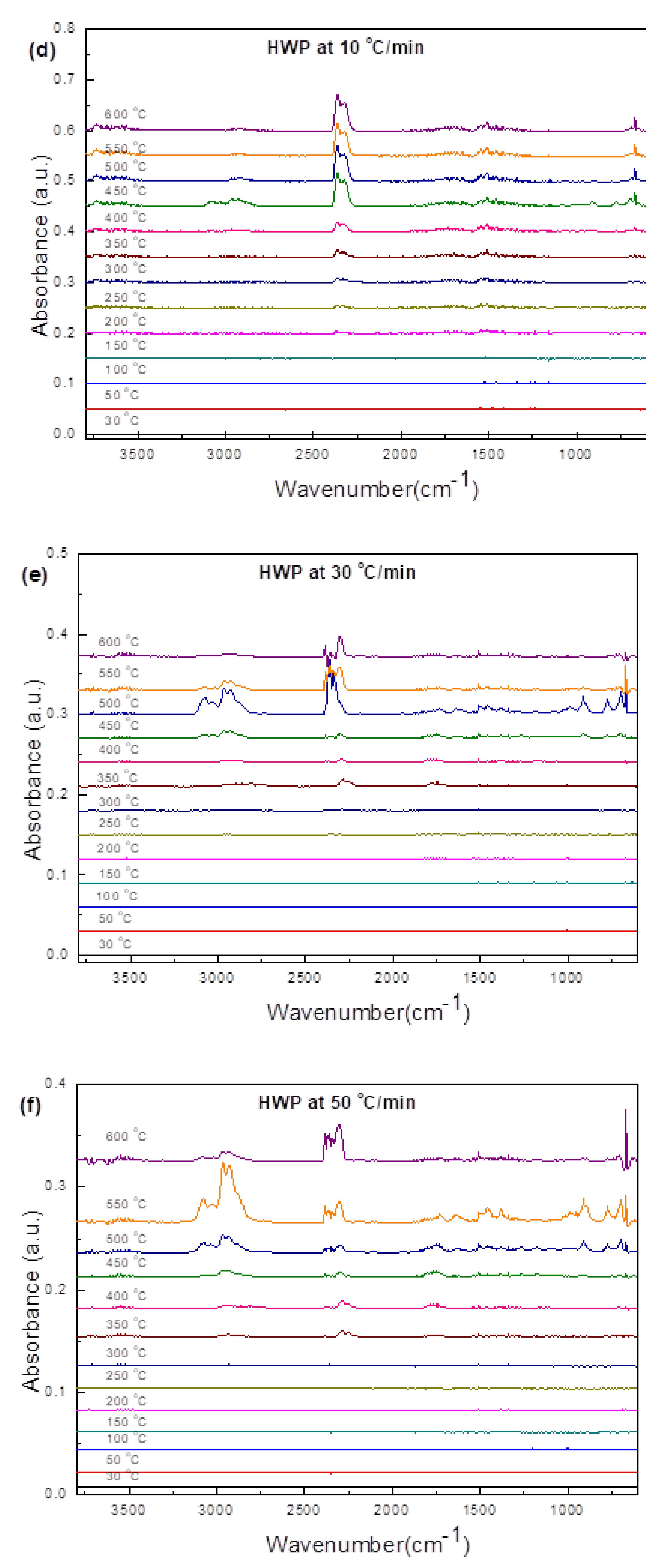
3.3.3. Hard White Plastic (HWP)
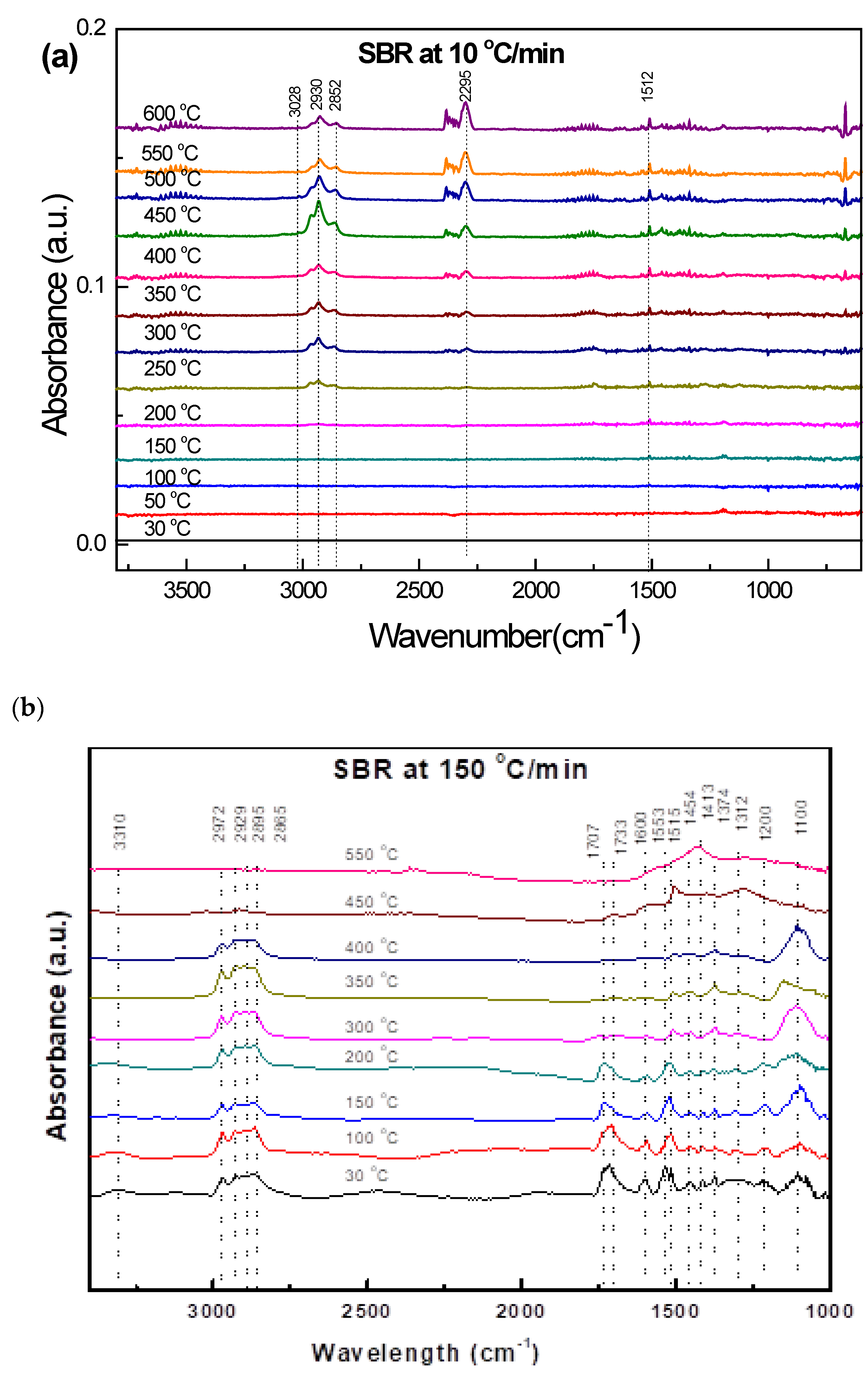
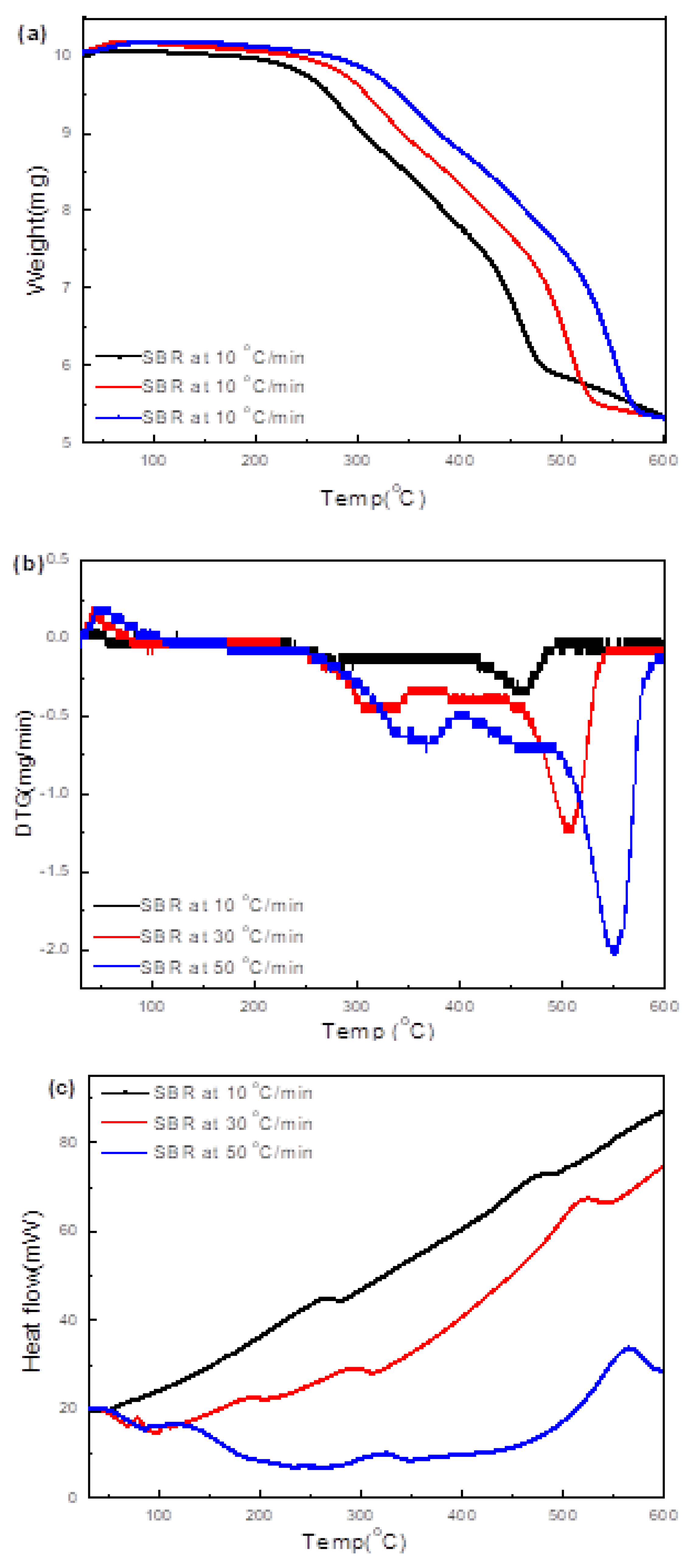
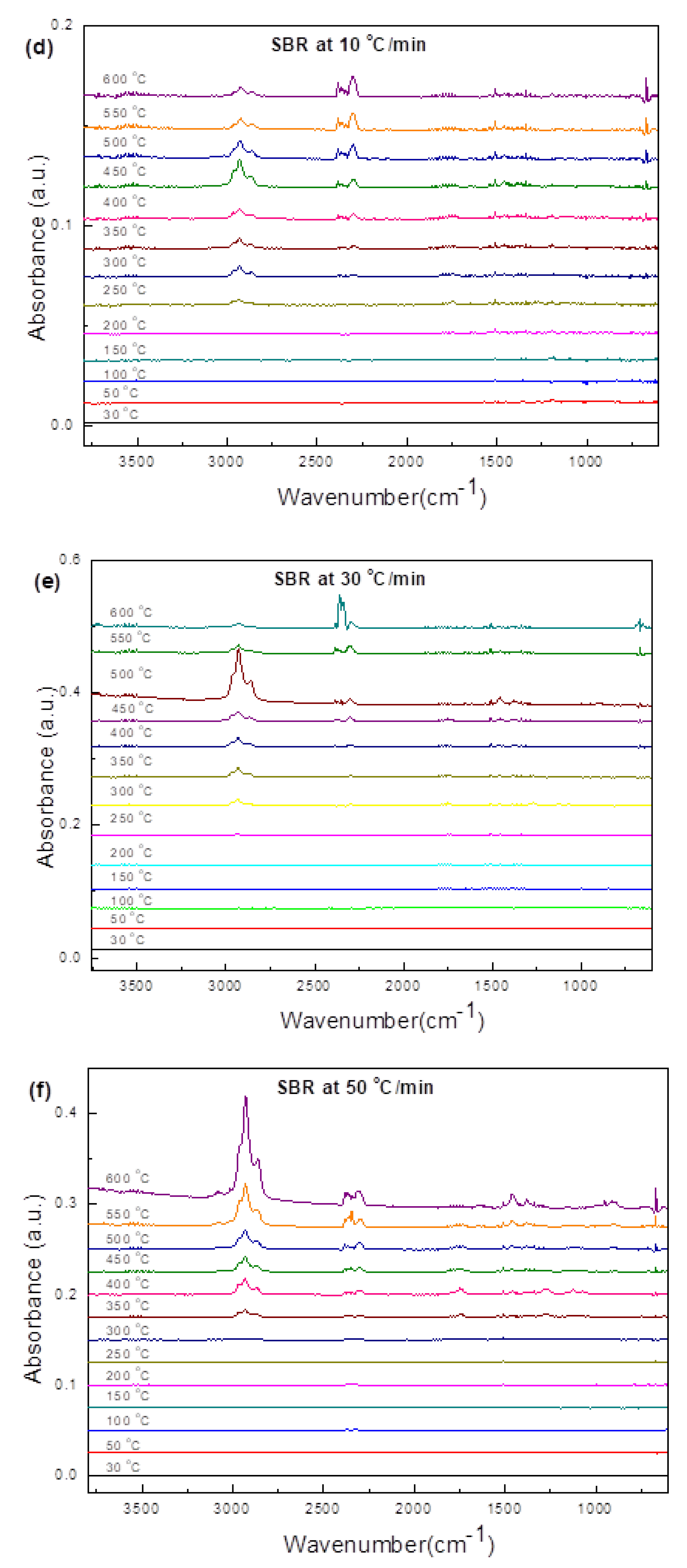
3.3.4. Soft Black Rubber (SBR)
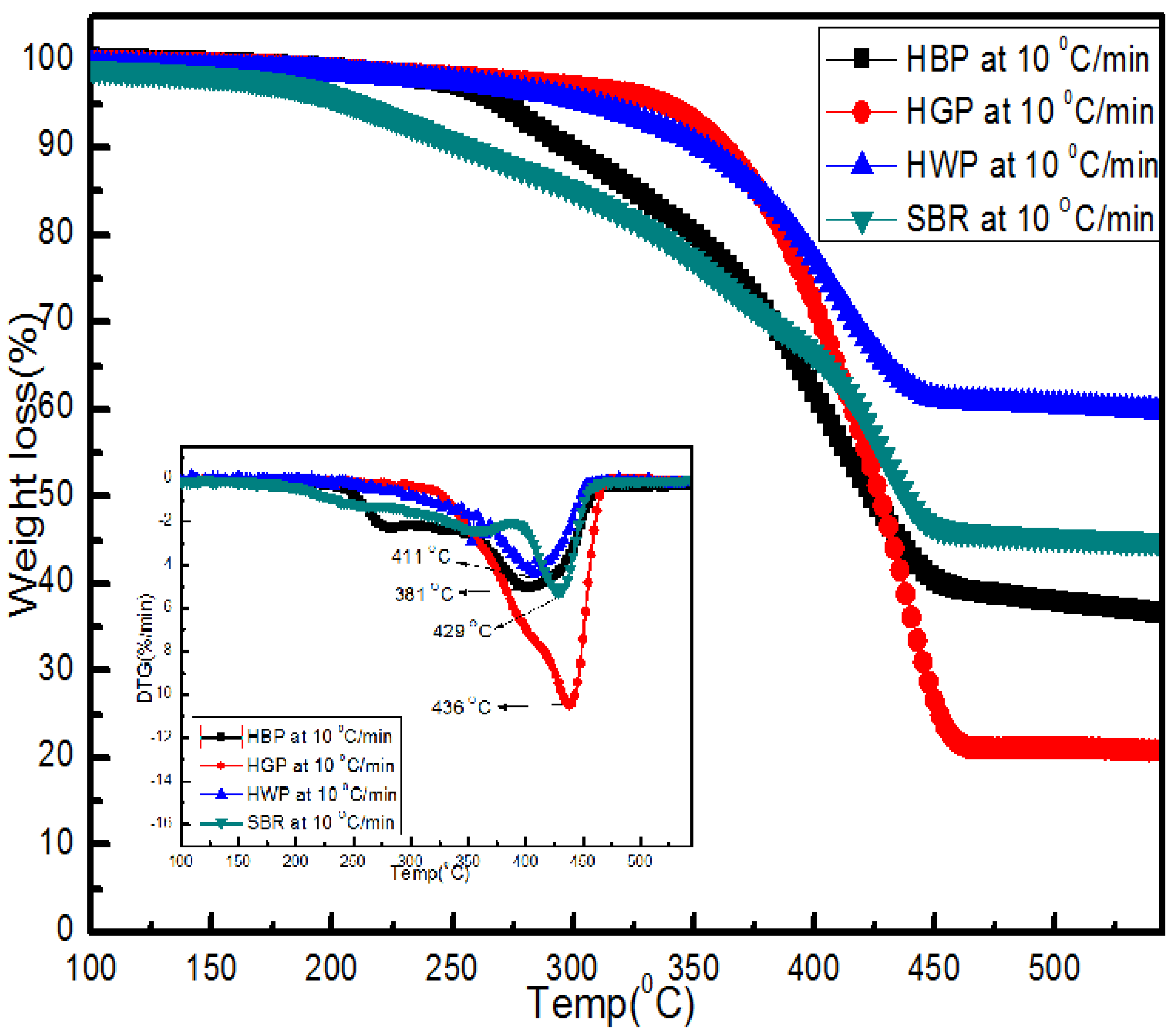
3.4. TG/DTG/DSC and Simultaneous IR Analysis of Evolved Gases
3.4.1. HBP
3.4.2. HGP
3.4.3. HWP
3.4.4. SBR
4. Conclusions
- (i)
- Morphology differs considerably among the different ASR component types and their chars, suggesting the difference in their chemical nature. The main matrix in each case was polymeric that pyrolysed with considerable char residue. There were considerable amounts of rod-shaped structures identified to be compounds such as oxides of Te, Sr, Si, As, Dy, Rn, Ti, and Nb (these thermally stable compounds do not transform during pyrolysis).
- (ii)
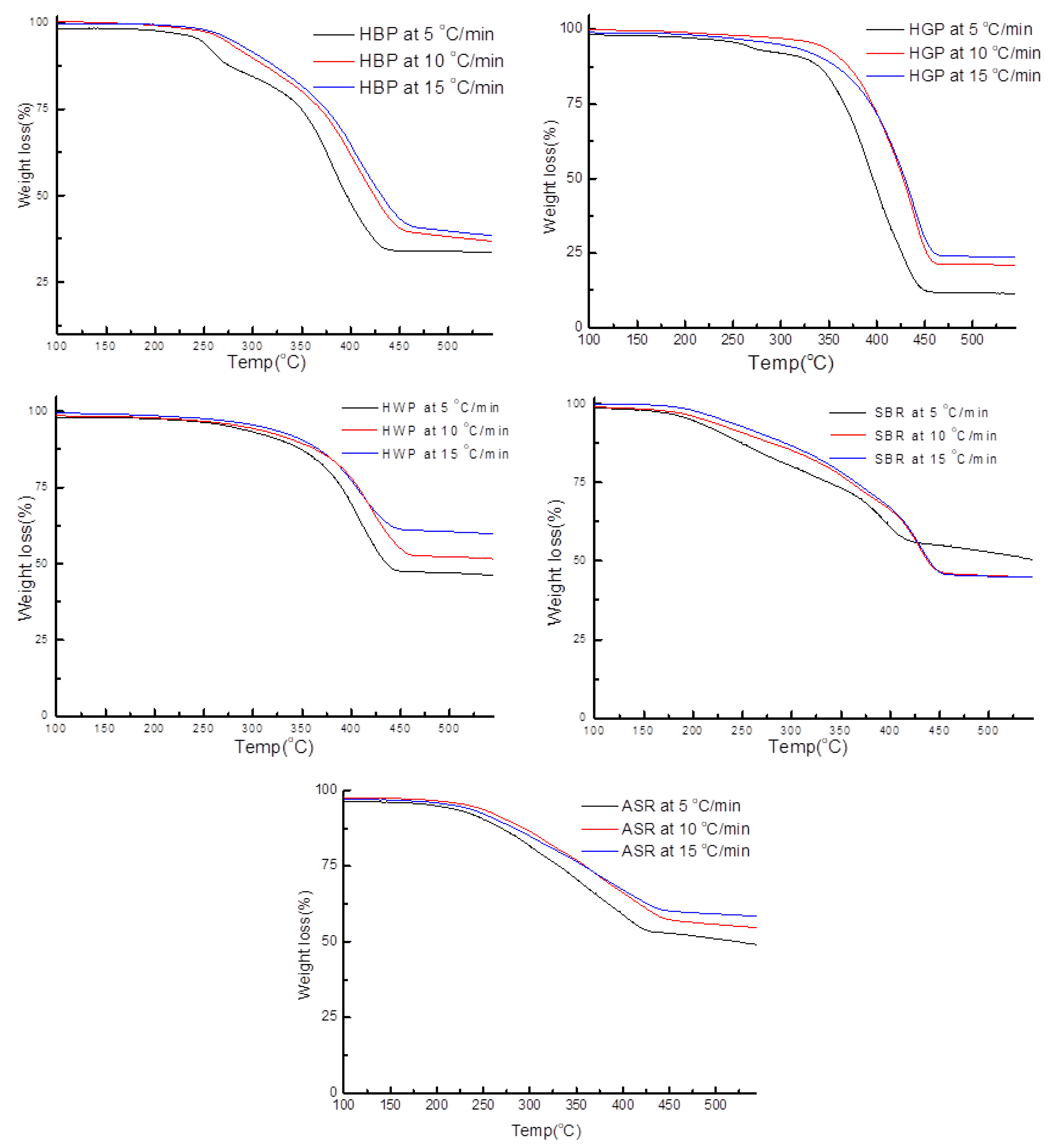
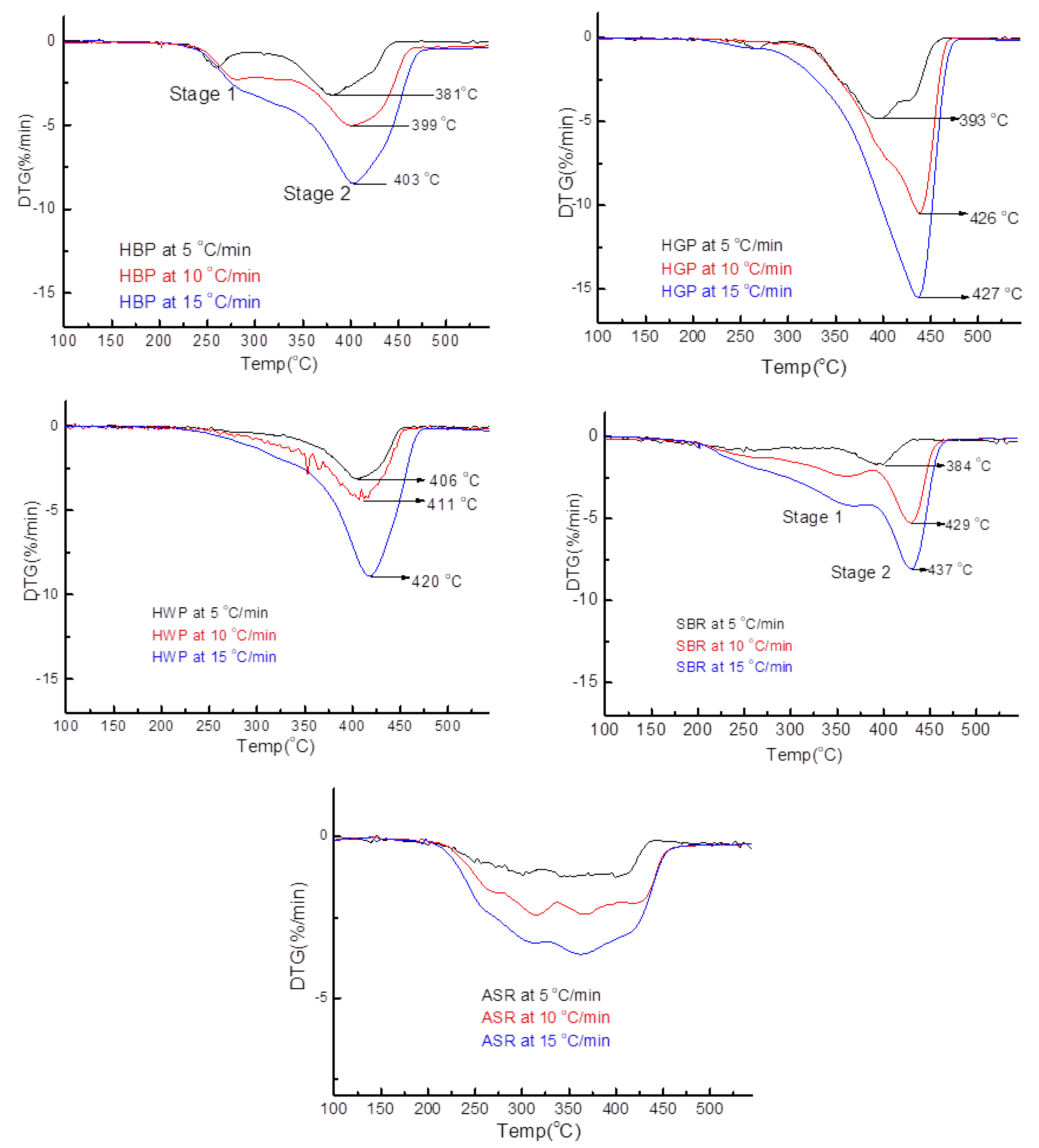
- TG/DTG, during heating to different temperatures (up to a maximum of 600 °C) at different heating rates, suggested either a single- or two-stage pyrolysis for different ASR components.
- (iii)
- Transformations in the first stage were generally attributed to the degradation of oxygen-containing functional groups, such as carbonyl, hydroxyl, or carboxyl groups (e.g., aldehydes and esters that are present in ASRs as impurities, additives, stabilizers, and/or oils) into carbon dioxide, methane, and water. Such transformations occurred at an elevated temperature, and the temperature range varied for HBP, HGP, HWP, and SBR.
- (iv)
- The second stage transformations occurred at higher temperatures, probably due to the thermal degradation of the polymer chain.
- (v)
- The TG/DTG/DSC and simultaneous gas-phase IR spectroscopy confirmed the evolution of at least some of the gases as described in (iii) above.
- (vi)
- In industrial practice, one would like to speed up the pyrolysis of ASRs into useful products such as oil by employing high heating rates. However, the synchrotron IR data presented in this study clearly demonstrate that a high heating rate (such as 150 °C/min) results in an incomplete conversion of ASRs unless necessary time is provided.
- (vii)
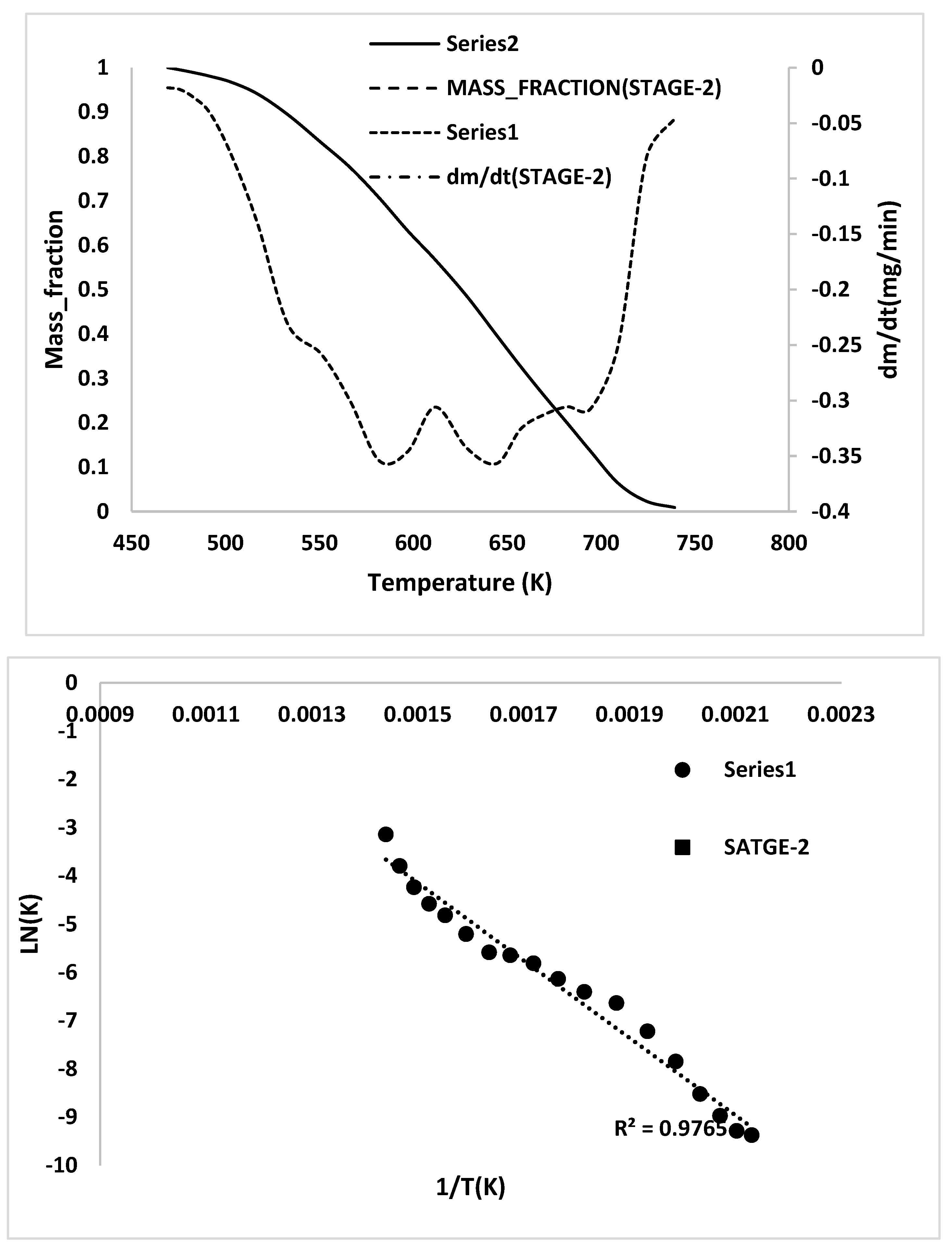
- To complement the information on the evolution of functional groups from ASR pyrolysis as a function of isothermal pyrolysis temperature and estimation of kinetic parameters based on multistage modelling, in situ transmission IR spectroscopy and TG analysis were used extensively. Thermogravimetric analysis shows that the maximum weight loss of ASR components occurred in the temperature range of 182–489 °C, at which 63.16, 82, 48.6, 51.76, 47.1% for the HBP, HGP, HWP, SBR, and the ASR mixture, respectively, was devolatilized. The linearized multistage kinetic model was applied to TGA data to obtain a good fit with a range (0.9513 < R2 < 0.9981) for all heating rates. Activation energy of different pyrolysis stages (24.98–124.94 kJ/mol) decreased with the increase in heating rate. The corresponding frequency factor ranged from 3.34 × 1013 to 5.68 × 101 mg−1/min. The reaction order for different stages in decomposition was in the range from 0.1 to 1.8.
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Appendix A

Appendix B

Appendix C

Appendix D

Appendix E

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Plastics | Stage-1 Devolatilization | Stage-2 Devolatilization |
|---|---|---|
| Hard black plastic (HDPE) 5 °C/min | Range: 212–293 °C Peak: 261 °C | Range: 312–455 °C Peak: 381 °C |
| 10 °C/min | Range: 211–302 °C Peak: 290 °C | Range: 355–470 °C Peak: 399 °C |
| 15 °C/min | Range: 213–309 °C Peak: 294 °C | Range: 332–482 °C Peak: 403 °C |
| Hard grey plastic (PP) 5 °C/min | Range: 241–287 °C Peak: 267 °C | Range: 299–469 °C Peak: 393 °C |
| 10 °C/min | Peak 1 Disappeared | Range: 230–481 °C Peak: 436 °C |
| 15 °C/min | Peak 1 Disappeared | Range: 212–486 °C Peak: 437 °C |
| Hard white plastic (PS) 5 °C/min | Range: 204–467 °C Peak: 406 °C | N/A |
| 10 °C/min | Range: 243–468 °C Peak: 411 °C | |
| 15 °C/min | Range: 246–489 °C Peak: 420 °C | |
| Soft black rubber (NR & SBR) 5 °C/min | Range: 145–297 °C Peak: 247 °C | Range: 341–433 °C Peak: 394 °C |
| 10 °C/min | Range: 148–386 °C Peak: 358 °C | Range: 391–478 °C Peak: 429 °C |
| 15 °C/min | Range: 160–385 °C Peak: 367 °C | Range: 390–482 °C Peak: 432 °C |
| Mixed ASR 5 °C/min | Range: 132–448 °C Peak: N/A | N/A |
| 10 °C/min | Range: 162–475 °C Peak: N/A | |
| 15 °C/min | Range: 166–478 °C Peak: N/A |
References
- Sakai, S.-i.; Yoshida, H.; Hiratsuka, J.; Vandecasteele, C.; Kohlmeyer, R.; Rotter, V.S.; Passarini, F.; Santini, A.; Peeler, M.; Li, J.; et al. An international comparative study of end-of-life vehicle (ELV) recycling systems. J. Mater. Cycles Waste Manag. 2014, 16, 1–20. [Google Scholar] [CrossRef]
- Davies, G. Materials for Automobile Bodies; Butterworth-Heinemann: Oxford, UK, 2012. [Google Scholar]
- Alonso, J.C.; Dose, J.; Fleischer, G.; Geraghty, K.; Greif, A.; Rodrigo, J.; Schmidt, W.-P. Electrical and electronic components in the automotive sector: Economic and environmental assessment. Int. J. Life Cycle Assess. 2007, 12, 328. [Google Scholar] [CrossRef]
- Goda, R. Use of Automotive Shredder Residue for the Nitro-Carburization of Steel: From Waste to Chemically-Bonded Ceramic Surface on Steel. Masters’s Thesis, UNSW Sydney, Sydney, Australia, 2016. [Google Scholar]
- Howard, G.T. Biodegradation of polyurethane: A review. Int. Biodeterior. Biodegrad. 2002, 49, 245–252. [Google Scholar] [CrossRef]
- Cossu, R.; Lai, T. Washing treatment of automotive shredder residue (ASR). Waste Manag. 2013, 33, 1770–1775. [Google Scholar] [CrossRef] [PubMed]
- Fiore, S.; Ruffino, B.; Zanetti, M.C. Automobile shredder residues in Italy: Characterization and valorization opportunities. Waste Manag. 2012, 32, 1548–1559. [Google Scholar] [CrossRef] [PubMed]
- Urbini, G.; Viotti, P.; Gavasci, R. Attenuation of methane, PAHs and VOCs in the soil covers of an automotive shredded residues landfill: A case study. J. Chem. Pharm. Res. 2014, 6, 618–625. [Google Scholar]
- Cossu, R.; Lai, T. Automotive shredder residue (ASR) management: An overview. Waste Manag. 2015, 45, 143–151. [Google Scholar] [CrossRef]
- Raboni, M.; Torretta, V.; Urbini, G.; Viotti, P. Automotive shredder residue: A survey of the hazardous organic micro-pollutants spectrum in landfill biogas. Waste Manag. Res. 2015, 33, 48–54. [Google Scholar] [CrossRef]
- Lee, H.-Y.; Oh, J.-K. A Study on the Shredding of End-of-Life Vehicles and Materials Separation. Geosystem Eng. 2003, 6, 100–105. [Google Scholar] [CrossRef]
- Vijayan, S.K.; Sahajwalla, V.; Bhattacharya, S. Insights into the options of energy and metal recovery from automotive shredder residue: A review. Resour. Conserv. Recycl. Adv. 2022, 15, 200097. [Google Scholar] [CrossRef]
- Simic, V.; Dimitrijevic, B. Modelling of automobile shredder residue recycling in the Japanese legislative context. Expert Syst. Appl. 2013, 40, 7159–7167. [Google Scholar] [CrossRef]
- Jody, B.; Daniels, E.; Duranceau, C.; Pomykala, J.; Spangenberger, J. End-of-Life Vehicle Recycling: State of the Art of Resource Recovery from Shredder Residue; Argonne National Laboratory (ANL): Lemont, IL, USA, 2011.
- Cossu, R.; Fiore, S.; Lai, T.; Luciano, A.; Mancini, G.; Ruffino, B.; Viotti, P.; Zanetti, M. Review of Italian experience on automotive shredder residue characterization and management. Waste Manag. 2014, 34, 1752–1762. [Google Scholar] [CrossRef] [PubMed]
- Zolezzi, M.; Nicolella, C.; Ferrara, S.; Iacobucci, C.; Rovatti, M. Conventional and fast pyrolysis of automobile shredder residues (ASR). Waste Manag. 2004, 24, 691–699. [Google Scholar] [CrossRef]
- De Marco, I.; Caballero, B.; Cabrero, M.; Laresgoiti, M.; Torres, A.; Chomon, M. Recycling of automobile shredder residues by means of pyrolysis. J. Anal. Appl. Pyrolysis 2007, 79, 403–408. [Google Scholar] [CrossRef]
- Chiarioni, A.; Reverberi, A.; El-Shaarawi, A.; Dovì, V. Modelling of an ASR countercurrent pyrolysis reactor with nonlinear kinetics. Appl. Therm. Eng. 2003, 23, 1847–1855. [Google Scholar] [CrossRef]
- Soustelle, M. Decomposition Reactions of Solids. In Handbook of Heterogenous Kinetics; John Wiley & Sons: Hoboken, NJ, USA, 2013; pp. 449–488. [Google Scholar]
- Burnham, A.K. Global Chemical Kinetics of Fossil Fuels: How to Model Maturation and Pyrolysis; Springer: Cham, Switzerland, 2017. [Google Scholar]
- Ni, F.; Chen, M. Research on ASR in China and its energy recycling with pyrolysis method. J. Mater. Cycles Waste Manag. 2015, 17, 107–117. [Google Scholar] [CrossRef]
- Hemati, S.; Hossain, R.; Sahajwalla, V. Selective thermal transformation of automotive shredder residues into high-value nano silicon carbide. Nanomaterials 2021, 11, 2781. [Google Scholar] [CrossRef]
- Sørum, L.; Grønli, M.; Hustad, J.E. Pyrolysis characteristics and kinetics of municipal solid wastes. Fuel 2001, 80, 1217–1227. [Google Scholar] [CrossRef]
- Yan, G.; Jing, X.; Wen, H.; Xiang, S. Thermal Cracking of Virgin and Waste Plastics of PP and LDPE in a Semibatch Reactor under Atmospheric Pressure. Energy Fuels 2015, 29, 2289–2298. [Google Scholar] [CrossRef]
- Senneca, O.; Chirone, R.; Masi, S.; Salatino, P. A Thermogravimetric Study of Nonfossil Solid Fuels. 1. Inert Pyrolysis. Energy Fuels 2002, 16, 653–660. [Google Scholar] [CrossRef]
- Aguado, R.; Gaisán, B.; Prieto, R.; Bilbao, J. Kinetics of polystyrene pyrolysis in a conical spouted bed reactor. Chem. Eng. J. 2003, 92, 91–99. [Google Scholar] [CrossRef]
- Bawn, C. Encyclopedia of Polymer Science and Engineering; Kroschwitz, J.I., Mark, H.F., Bikales, N., Overberger, C.G., Menges, G., Eds.; John Wiley and Sons: New York, NY, USA, 1985; p. 906. ISBN 0-471-89540-7. [Google Scholar]
- Williams, P.T.; Besler, S. Pyrolysis-thermogravimetric analysis of tyres and tyre components. Fuel 1995, 74, 1277–1283. [Google Scholar] [CrossRef]
- Acevedo, B.; Fernández, A.; Barriocanal, C. Identification of polymers in waste tyre reinforcing fibre by thermal analysis and pyrolysis. J. Anal. Appl. Pyrolysis 2015, 111, 224–232. [Google Scholar] [CrossRef]
- Moldoveanu, S.C. Analytical Pyrolysis of Natural Organic Polymers; Elsevier: Amsterdam, The Netherlands, 1998; Volume 20. [Google Scholar]
- Miskolczi, N.; Bartha, L.; Deák, G.; Jóver, B. Thermal degradation of municipal plastic waste for production of fuel-like hydrocarbons. Polym. Degrad. Stab. 2004, 86, 357–366. [Google Scholar] [CrossRef]
- Colthup, N. Introduction to Infrared and Raman Spectroscopy; Elsevier: Amsterdam, The Netherlands, 2012. [Google Scholar]
- Miandad, R.; Barakat, M.; Aburiazaiza, A.S.; Rehan, M.; Ismail, I.; Nizami, A. Effect of plastic waste types on pyrolysis liquid oil. Int. Biodeterior. Biodegrad. 2017, 119, 239–252. [Google Scholar] [CrossRef]
- Stuart, B.H. Polymers. In Infrared Spectroscopy: Fundamentals and Applications; John Wiley & Sons, Ltd.: Hoboken, NJ, USA, 2005; pp. 113–136. [Google Scholar] [CrossRef]
- Stuart, B.H. Spectral Analysis. In Infrared Spectroscopy: Fundamentals and Applications; John Wiley & Sons, Ltd.: Hoboken, NJ, USA, 2005; pp. 45–70. [Google Scholar] [CrossRef]
- Tidjani, A. Comparison of formation of oxidation products during photo-oxidation of linear low density polyethylene under different natural and accelerated weathering conditions. Polym. Degrad. Stab. 2000, 68, 465–469. [Google Scholar] [CrossRef]
- Mamoor, G.; Shahid, W.; Mushtaq, A.; Amjad, U.; Mehmood, U. Recycling of mixed plastics waste containing polyethylene, polyvinylchloride and polyethylene terephthalate. Chem. Eng. Res. Bull. 2013, 16, 25–32. [Google Scholar] [CrossRef]
- Gao, F. Pyrolysis of Waste Plastics into Fuels. Ph.D. Thesis, University of Canterbury, Christchurch, New Zealand, 2010. [Google Scholar]
- Seidelt, S.; Müller-Hagedorn, M.; Bockhorn, H. Description of tire pyrolysis by thermal degradation behaviour of main components. J. Anal. Appl. Pyrolysis 2006, 75, 11–18. [Google Scholar] [CrossRef]
- Larsen, M.B.; Schultz, L.; Glarborg, P.; Skaarup-Jensen, L.; Dam-Johansen, K.; Frandsen, F.; Henriksen, U. Devolatilization characteristics of large particles of tyre rubber under combustion conditions. Fuel 2006, 85, 1335–1345. [Google Scholar] [CrossRef]






















| Proximate Analysis (wt.%) | Ultimate Analysis (wt.%) | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Sample | Tmax °C | VM | A | M | FC | C | H | N | S | Odiff | GCV (MJ/kg) |
| HBP | 290, 399 | 62.1 | 17.0 | 0.8 | 20.0 | 61.9 | 6.4 | 2.7 | 0.3 | 28.6 | 24.8 |
| HGP | 436 | 82.0 | 0.3 | 0.1 | 17.5 | 69.7 | 8.6 | 3.4 | 0.1 | 18.4 | 32.6 |
| HWP | 411 | 48.6 | 28.3 | 0.8 | 22.2 | 49.0 | 5.5 | 1.2 | 0.2 | 43.9 | 17.5 |
| SBR | 358, 429 | 51.7 | 23.5 | 4.3 | 20.3 | 65.0 | 7.2 | 0.5 | 0.8 | 26.4 | 27.6 |
| ASR | 162–475 | 47.1 | 35.5 | 3.6 | 13.8 | - | - | - | - | - | - |
| Plastics | Wt.% |
|---|---|
| Polypropylene | 39 |
| Polyethylene | 28 |
| Polycarbonate | 12 |
| Polyurethane | 5 |
| other | 16 |
| Polymer Type | Temp Range (°C) | Peak Temp (°C) | References |
|---|---|---|---|
| HDPE (10 °C/min) | 350–380 | 480 | [23] |
| LDPE (10 °C/min) | 380–500 | 470 | [24] |
| PE (20 °C/min) | 400–520 | 480 | [25] |
| LDPE (20 °C/min) | 410–515 | 491 | [26] |
| PP (10 °C/min) | 328–410 | 464 | [27] |
| NR (10 °C/min) | - | 378 | [28,29] |
| SBR (10 °C/min) | - | 447 | [28,29] |
| BR (10 °C/min) | - | 467 | [28,29] |
| BR + SBR (10 °C/min) | - | 427 | [28,29] |
| 10 °C/min, Current Data, Wave Number (cm−1) | 150 °C/min, Current Data, Wave Number (cm−1) | Functional Groups | Wave Number (cm−1) | Reference |
|---|---|---|---|---|
| 3293 | 3300 | Amide N--H stretch | 3550–3250 | [32] |
| 2958 | 2980 | Methylene (as) C--H stretching | 2936–2843 | [32] |
| 2962 | 2918 | Methylene (as) C-H stretching | 2936–2843 | [32] |
| 2855 | 2800 | Methylene (s) C-H stretching, C--H stretch in N-CH2 | 2936–2843, 2820–2760 | [32] |
| 1640 | 1640 | Olefinic -C=C-, Amide C=O stretching | 1680–1600 1695–1630 | [32] |
| 1534 | - | Amide II N-H bending and C-N stretching | 1565–1508 | [32] |
| 1452 | 1470 | CH2-CH2-[(NH-C-H bending | 1475–1445 | [32] |
| 1361 | 1382 | Methyl C-H bending | 1390–1370 | [32] |
| - | 1282 | Amide III C-N stretching -NH bending | 1300–1000 | [32] |
| - | 1233 | Amide III C-N stretching -NH bending | 1300–1000 | [32] |
| 1150 | CC-H (s) bending/CH2 twisting | - | [32] | |
| 996 | 999 | C-C stretching and bending | 1000–800 | [32] |
| 10 °C/min, Current Data, Wave Numbers (cm−1) | 150 °C/min, Current Data, Wave Numbers (cm−1) | Functional Groups | Wave Number (cm−1) | References |
|---|---|---|---|---|
| 2954 | 2961 | Methyl (s) C-H stretching | 2972–2862 | [32,33,34] |
| 2914 | 2916 | Methylene (s) C-H stretching | 2936–2843 | [32,33,34] |
| 2875 | 2868 | Methyl (s) C-H stretching | 2972–2862 | [32,33,34] |
| 2848 | 2872 | Methyl (s) C-H stretching | 2972–2862 | [32,33,34] |
| 1456 | 1459 | Methylene C-H bending O-H bending | 1475–1445 1440–1330 | [32,33,34] |
| 1373 | 1377 | Methyl C-H bending O-H bending | 1390–1370 1390–1310 | [32,34,35] |
| 1160 | 1167–1043 | Methylene (s) C-H deformation C-O stretch | 1250–900 1150–1085 | [34] |
| - | 997 | C-C stretching, bending and rocking | 1000–800 | [32,33,36] |
| 1000 | 974 | C-C stretching, bending and rocking | 1000–800 | [32,33,36] |
| 10 °C/min, Current Data, Wave Numbers (cm−1) | 150 °C/min, Current Data, Wave Numbers (cm−1) | Functional Group | Wave Number (cm−1) | References |
|---|---|---|---|---|
| 3067–3024 | 3061–2025 | Aromatic C-H stretching | 3100–3000 | [32,34] |
| 2935–2851 | 2938–2853 | Methylene (s) C-H stretching | 2936–2843 | |
| 1873–1734 | 1751–1701 | Overtone bands mono-substituted benzene rings | 2000–1700 | [34] |
| Carbonyl C=O stretching | 1760–1700 | [32,34] | ||
| 1597–1490 | 1600–1495 1530–1515 | Aromatic C=C stretching and methyl C--H bending Carbonyl C=O stretching | 1600–1430 | [32,33,34,37] |
| 1367 | 1305 | Methylene C-H wagging | 1390–1370 | [32,33,34] |
| 1188–1026 | 1139–1047 | Aromatic C-H bending | 1250–900 1275–1000 | [34] |
| 150 °C/min, Current Data, Wave numbers (cm−1) | Functional Groups | Wave Number (cm−1) | References |
|---|---|---|---|
| 3310 | O-H stretching | 3550–3200 | [32] |
| 2972–2885 | Methylene (s) C-H stretching | 2936–2843 | [32] |
| 1733–1707 | Carbonyl C=O stretching | 1760–1700 | [32,34] |
| 1600 | C=C stretching | 1600–1680 | [32,34] |
| 1533–1515 1454–1374 1312–1100 | Carbonyl C=O stretching Methylene C-H bending and wagging Vinyl CCH bending and twisting | - 1470–1450 - | [34] [34] [34] |
| Hard black plastic (PA) 5 °C/min | n | 1 | n | 0.3 |
| E | 124.94 | E | 85.99 | |
| A | 3.34 × 1013 | A | 1.06 × 107 | |
| 10 °C/min | n | 0.9 | n | 0.6 |
| E | 114.52 | E | 54.73 | |
| A | 3.83 × 1010 | A | 6.20 × 103 | |
| 15 °C/min | n | 0.3 | n | 0.6 |
| E | 106.43 | E | 35.73 | |
| A | 1.19 × 1010 | A | 4.72 × 102 | |
| Hard grey plastic (PP) 5 °C/min | n | 1.1 | n | 1 |
| E | 76.20 | E | 126.47 | |
| A | 2.13 × 106 | A | 1.00 × 109 | |
| 10 °C/min | n | 1 | n | N/A |
| E | 71.20 | E | ||
| A | 4.32 × 104 | A | ||
| 15 °C/min | n | 0.1 | n | N/A |
| E | 71.23 | E | ||
| A | 1.86 × 105 | A | ||
| Hard white plastic (PS) 5 °C/min | n | 1.1 | n | N/A |
| E | 67.00 | E | ||
| A | 3.52 × 1011 | A | ||
| 10 °C/min | n | 1 | n | N/A |
| E | 59.64 | E | ||
| A | 2.14 × 104 | A | ||
| 15 °C/min | n | 0.1 | n | N/A |
| E | 51.16 | E | ||
| A | 5.20 × 103 | A | ||
| Soft black rubber (XNBR) 5 °C/min | n | 0.1 | n | 0.8 |
| E | 62.08 | E | 124.27 | |
| A | 3.77 × 102 | A | 2.60 × 109 | |
| 10 °C/min | n | 0.2 | n | 0.6 |
| E | 35.77 | E | 110.11 | |
| A | 4.43 × 102 | A | 3.25 × 1011 | |
| 15 °C/min | n | 0.1 | n | 0.6 |
| E | 24.98 | E | 121.79 | |
| A | 5.68 × 101 | A | 1.03 × 109 | |
| Mixed ASR 5 °C/min | n | 1.7 | n | N/A |
| E | 92.24 | E | ||
| A | 2.67 × 104 | A | ||
| 10 °C/min | n | 1.7 | n | N/A |
| E | 78.66 | E | ||
| A | 2.67 × 104 | A | ||
| 15 °C/min | n | 1.8 | n | N/A |
| E | 71.18 | E | ||
| A | 1.29 × 105 | A |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kohli, I.; Srivatsa, S.C.; Das, O.; Devasahayam, S.; Singh Raman, R.K.; Bhattacharya, S. Pyrolysis of Automotive Shredder Residue (ASR): Thermogravimetry, In-Situ Synchrotron IR and Gas-Phase IR of Polymeric Components. Polymers 2023, 15, 3650. https://doi.org/10.3390/polym15173650
Kohli I, Srivatsa SC, Das O, Devasahayam S, Singh Raman RK, Bhattacharya S. Pyrolysis of Automotive Shredder Residue (ASR): Thermogravimetry, In-Situ Synchrotron IR and Gas-Phase IR of Polymeric Components. Polymers. 2023; 15(17):3650. https://doi.org/10.3390/polym15173650
Chicago/Turabian StyleKohli, Isha, Srikanth Chakravartula Srivatsa, Oisik Das, Sheila Devasahayam, R. K. Singh Raman, and Sankar Bhattacharya. 2023. "Pyrolysis of Automotive Shredder Residue (ASR): Thermogravimetry, In-Situ Synchrotron IR and Gas-Phase IR of Polymeric Components" Polymers 15, no. 17: 3650. https://doi.org/10.3390/polym15173650