
Optimization of Approaches to Analysis of Lignin by Thermal Decomposition

Abstract
:1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Isolation of Lignins
2.2.1. Milled Wood Lignin
2.2.2. Alkaline Ethanol Lignin
2.2.3. Dioxanelignin
2.3. Analytical Methods
2.3.1. NMR Spectroscopy
2.3.2. Thermal Desorption
2.3.3. Analytical Pyrolysis
3. Results and Discussion
3.1. Optimization of TD–GC/MS Analysis Parameters for Lignin Decomposition
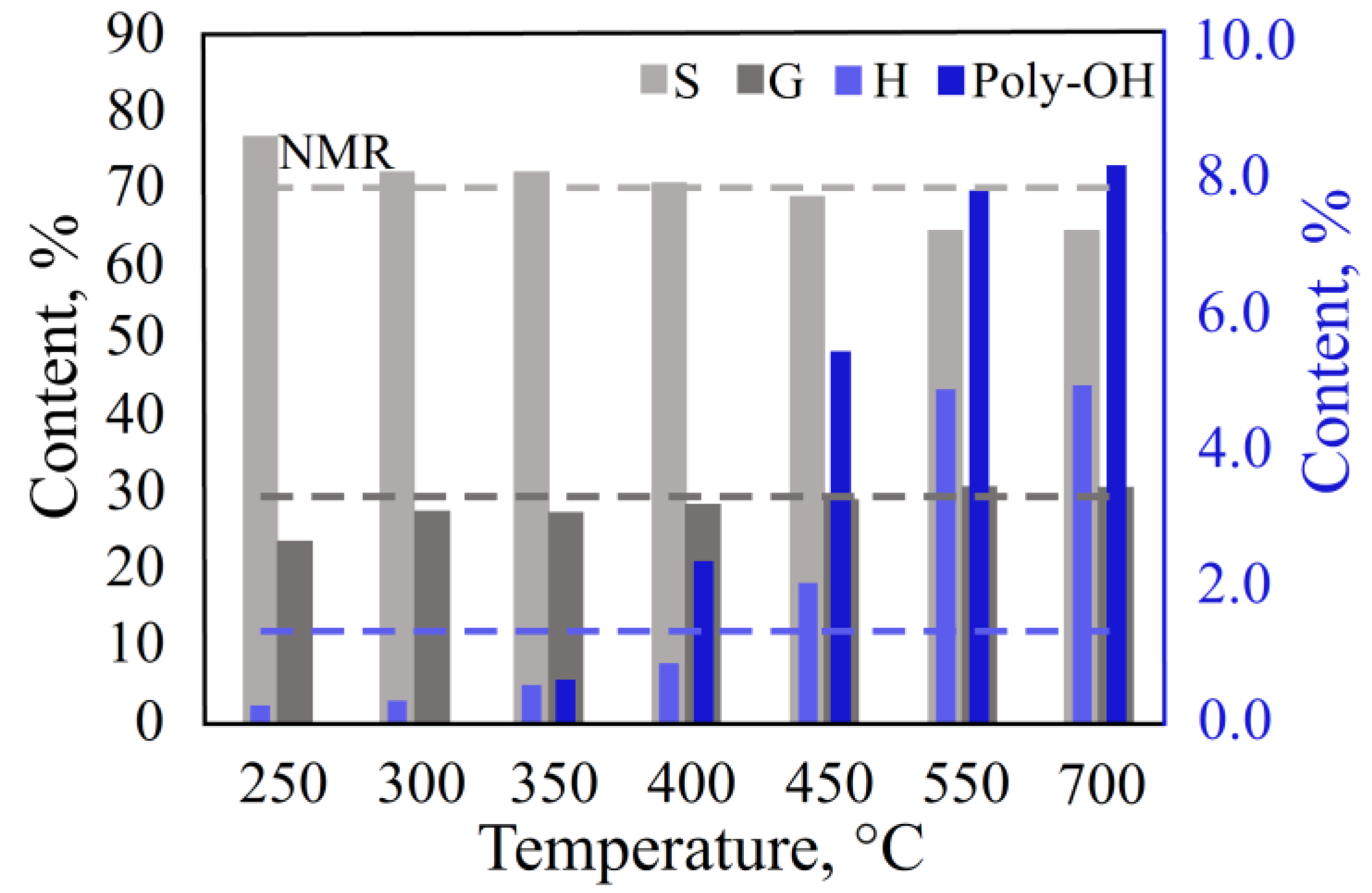
3.1.1. Effect of Temperature on the Thermal Decomposition
3.1.2. Effect of Sample Weight
3.1.3. Effect of TD Duration
3.2. Optimization of Py–GC/MS Analysis Parameters for Lignin Decomposition
3.2.1. Heating Rate in the Double-Shot Mode
3.2.2. Effect of Sample Weight in the DS Mode
3.2.3. Effect of Temperature in the DS Mode
3.2.4. Effect of Temperature in the Single-Shot Mode
3.3. Application of the Specified Thermal Decomposition Conditions for Various Lignin Preparations
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Hu, J.; Zhang, Q.; Lee, D.J. Kraft lignin biorefinery: A perspective. Bioresour. Technol. 2018, 247, 1181–1183. [Google Scholar] [CrossRef] [PubMed]
- Yuan, T.Q.; Xu, F.; Sun, R.C. Role of lignin in a biorefinery: Separation characterization and valorization. J. Chem. Technol. Biotechnol. 2013, 88, 346–352. [Google Scholar] [CrossRef]
- Sun, R.C. Lignin source and structural characterization. ChemSusChem 2020, 13, 4385–4393. [Google Scholar] [CrossRef] [PubMed]
- Balakshin, M.Y.; Capanema, E.A.; Sulaeva, I.; Schlee, P.; Huang, Z.; Feng, M.; Borghei, M.; Rojas, O.J.; Potthast, A.; Rosenau, T. New opportunities in the valorization of technical lignins. ChemSusChem 2021, 14, 1016–1036. [Google Scholar] [CrossRef]
- Boerjan, W.; Ralph, J.; Baucher, M. Lignin biosynthesis. Annu. Rev. Plant Biol. 2003, 54, 519–546. [Google Scholar] [CrossRef]
- Anderson, E.M.; Stone, M.L.; Katahira, R.; Reed, M.; Muchero, W.; Ramirez, K.J.; Beckham, G.T.; Román-Leshkov, Y. Differences in S/G ratio in natural poplar variants do not predict catalytic depolymerization monomer yields. Nat. Commun. 2019, 10, 2033. [Google Scholar] [CrossRef] [Green Version]
- Li, C.; Zhao, X.; Wang, A.; Huber, G.W.; Zhang, T. Catalytic transformation of lignin for the production of chemicals and fuels. Chem. Rev. 2015, 115, 11559–11624. [Google Scholar] [CrossRef]
- Del Rio, J.C.; Gutiérrez, A.; Rodríguez, I.M.; Ibarra, D.; Martinez, A.T. Composition of non-woody plant lignins and cinnamic acids by Py-GC/MS, Py/TMAH and FT-IR. J. Anal. Appl. Pyrolysis 2007, 79, 39–46. [Google Scholar] [CrossRef]
- Paulsen Thoresen, P.; Lange, H.; Crestini, C.; Rova, U.; Matsakas, L.; Christakopoulos, P. Characterization of organosolv birch lignins: Toward application-specific lignin production. ACS Omega 2021, 6, 4374–4385. [Google Scholar] [CrossRef]
- Martín-Sampedro, R.; Santos, J.I.; Fillat, Ú.; Wicklein, B.; Eugenio, M.E.; Ibarra, D. Characterization of lignins from Populus alba L. generated as by-products in different transformation processes: Kraft pulping, organosolv and acid hydrolysis. Int. J. Biol. Macromol. 2019, 126, 18–29. [Google Scholar] [CrossRef]
- Lupoi, J.S.; Singh, S.; Parthasarathi, R.; Simmons, B.A.; Henry, R.J. Recent innovations in analytical methods for the qualitative and quantitative assessment of lignin. Renew. Sust. Energ. Rev. 2015, 49, 871–906. [Google Scholar] [CrossRef] [Green Version]
- Shao, Z.; Fu, Y.; Wang, P.; Zhang, Y.; Qin, M.; Li, X.; Zhang, F. Modification of the aspen lignin structure during integrated fractionation process of autohydrolysis and formic acid delignification. Int. J. Biol. Macromol. 2018, 165, 1727–1737. [Google Scholar] [CrossRef] [PubMed]
- Ona, T.; Sonoda, T.; Ito, K.; Shibatal, M.; Katayama, T.; Kato, T.; Ootake, Y. Non-destructive determination of lignin syringyl/guaiacyl monomeric composition in native wood by Fourier transform Raman spectroscopy. J. Wood Chem. Technol. 1998, 18, 43–51. [Google Scholar] [CrossRef]
- Balakshin, M.; Capanema, E.A.; Zhu, X.; Sulaeva, I.; Potthast, A.; Rosenau, T.; Rojas, O.J. Spruce milled wood lignin: Linear, branched or cross-linked? Green Chem. 2020, 22, 3985–4001. [Google Scholar] [CrossRef]
- Zeng, J.; Helms, G.L.; Gao, X.; Chen, S. Quantification of wheat straw lignin structure by comprehensive NMR analysis. J. Agric. Food Chem. 2013, 61, 10848–10857. [Google Scholar] [CrossRef]
- Xu, G.; Shi, Z.; Zhao, Y.; Deng, J.; Dong, M.; Liu, C.; Murugadoss, V.; Mai, X.; Guo, Z. Structural characterization of lignin and its carbohydrate complexes isolated from bamboo (Dendrocalamus sinicus). Int. J. Biol. Macromol. 2019, 126, 376–384. [Google Scholar] [CrossRef]
- Kumar, A.; Biswas, B.; Saini, K.; Kumar, A.; Kumar, J.; Krishna, B.B.; Bhaskar, T. Py-GC/MS study of prot lignin with cobalt impregnated titania, ceria and zirconia catalysts. Renew. Energ. 2021, 172, 121–129. [Google Scholar] [CrossRef]
- Branco, D.G.; Santiago, C.; Lourenco, A.; Cabrita, L.; Evtuguin, D.V. Structural Features of Cork Dioxane Lignin from Quercus suber L. J. Agric. Food Chem. 2021, 69, 8555–8564. [Google Scholar] [CrossRef]
- Rencoret, J.; Gutiérrez, A.; Marques, G.; José, C.; Tobimatsu, Y.; Lam, P.Y.; Pérez-Boada, M.; Ruiz-Dueñas, F.J.; Barrasa, M.; Martínez, A.T. New insights on structures forming the lignin-like fractions of ancestral plants. Front. Plant Sci. 2021, 12, 740923. [Google Scholar] [CrossRef]
- Reeves III, J.B.; Galletti, G.C. Use of pyrolysis—Gas chromatography/mass spectrometry in the study of lignin assays. J. Anal. Appl. Pyrolysis 1993, 24, 243–255. [Google Scholar] [CrossRef]
- Kawamoto, H. Lignin pyrolysis reactions. J. Wood Sci. 2017, 63, 117–132. [Google Scholar] [CrossRef] [Green Version]
- Izaguirre, N.; Robles, E.; Llano-Ponte, R.; Labidi, J.; Erdocia, X. Fine-tune of lignin properties by its fractionation with a sequential organic solvent extraction. Ind. Crops Prod. 2022, 175, 114251. [Google Scholar] [CrossRef]
- Sluiter, A.; Hames, B.; Ruiz, R.; Scarlata, C.; Sluiter, J.; Templeton, D.; Crocker, D. Determination of Structural Carbohydrates and Lignin in Biomass. Lab. Anal. Proced. 2008, 1617, 1–16. [Google Scholar]
- Bjorkman, A. Studies on finely divided wood. Part I. Extraction of lignin with neutral solvents. Sven. Papperstidn. 1956, 59, 477–485. [Google Scholar]
- Navaee-Ardeh, S.; Mohammadi-Rovshandeh, J.; Pourjoozi, M. Influence of rice straw cooking conditions in the soda–ethanol–water pulping on the mechanical properties of produced paper sheets. Bioresour. Technol. 2004, 92, 65–69. [Google Scholar] [CrossRef] [PubMed]
- Popova, Y.A.; Shestakov, S.L.; Belesov, A.V.; Pikovskoi, I.I.; Kozhevnikov, A.Y. Comprehensive analysis of the chemical structure of lignin from raspberry stalks (Rubus idaeus L.). Int. J. Biol. Macromol. 2020, 164, 3814–3822. [Google Scholar] [CrossRef] [PubMed]
- Tarasov, D.; Schlee, P.; Pranovich, A.; Moreno, A.; Wang, L.; Rigo, D.; Sipponen, M.H.; Xu, C.; Balakshin, M. AqSO biorefinery: A green and parameter-controlled process for the production of lignin–carbohydrate hybrid materials. Green Chem. 2022, 24, 6639–6656. [Google Scholar] [CrossRef]
- Del Río, J.C.; Rencoret, J.; Prinsen, P.; Martínez, Á.T.; Ralph, J.; Gutiérrez, A. Structural characterization of wheat straw lignin as revealed by analytical pyrolysis, 2D-NMR, and reductive cleavage methods. J. Agric. Food Chem. 2012, 60, 5922–5935. [Google Scholar] [CrossRef] [Green Version]
- Balakshin, M.; Capanema, E.; Gracz, H.; Chang, H.M.; Jameel, H. Quantification of lignin–carbohydrate linkages with high-resolution NMR spectroscopy. Planta 2011, 233, 1097–1110. [Google Scholar] [CrossRef]
- Ralph, J.; Landucci, L. NMR of Lignins. In Lignin and Lignans; CRC Press: Boca Raton, FL, USA, 2010. [Google Scholar]
- Capanema, E.; Balakshin, M.; Katahira, R.; Chang, H.M.; Jameel, H. How well do MWL and CEL preparations represent the whole hardwood lignin? J. Wood Chem. Technol. 2015, 35, 17–26. [Google Scholar] [CrossRef]
- Ralph, J.; Hatfield, D. Pyrolysis-GC–MS characterization of forage materials. J. Agric. Food Chem. 1991, 39, 1426–1437. [Google Scholar] [CrossRef]
- Meier, D.; Faix, O. Pyrolysis-gas chromatography-mass spectrometry of lignin. In Methods in Lignin Chemistry; Lin, S.Y., Dence, C.W., Eds.; Springer: Berlin/Heidelberg, Germany, 1992; pp. 177–199. [Google Scholar]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Parameter | Py–GC/MS | TD–GC/MS | |
|---|---|---|---|
| Single Shot | Double Shot | ||
| Final temperature, °C | 350 | 400 | 200–250 |
| Heating rate, °C/min | – | 120 | – |
| Process duration, min | 1 | 3 | 30 |
| Sample weight, mg | 0.2–0.4 | 0.2–0.4 | 1.5–3.0 |
| Unit 1,2 | Py–GC/MS | TD–GC/MS | HSQC NMR | |
|---|---|---|---|---|
| Single Shot | Double Shot | |||
| SMWL | 70.2 | 70.5 | 67.6 | 69.8 |
| SEtOH | 67.8 | 70.4 | 75.9 | 72.6 |
| SDL | 69.1 | 70.7 | 66.5 | 71.4 |
| GMWL | 29.4 | 28.7 | 31.8 | 29.5 |
| GEtOH | 31.5 | 28.7 | 23.2 | 26.6 |
| GDL | 30.4 | 29.1 | 33.5 | 27.9 |
| HMWL | 0.4 | 0.8 | 0.6 | 1.3 |
| HEtOH | 0.8 | 0.9 | 0.9 | 1.6 |
| HDL | 0.3 | 0.3 | 0.0 | 1.5 |
| Poly-OHMWL | 4.6 | 2.4 | 0.2 | - |
| Poly-OHEtOH | 1.2 | 3.4 | 0.0 | - |
| Poly-OHDL | 2.3 | 1.9 | 0.0 | - |
| Unit | Py–GC/MS | TD–GC/MS | |
|---|---|---|---|
| Single Shot | Double Shot | ||
| S | 2.50 | 1.20 | 3.47 |
| G | 2.50 | 1.37 | 3.77 |
| H | 0.97 | 0.80 | 0.97 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pokryshkin, S.; Sypalova, Y.; Ivahnov, A.; Kozhevnikov, A. Optimization of Approaches to Analysis of Lignin by Thermal Decomposition. Polymers 2023, 15, 2861. https://doi.org/10.3390/polym15132861
Pokryshkin S, Sypalova Y, Ivahnov A, Kozhevnikov A. Optimization of Approaches to Analysis of Lignin by Thermal Decomposition. Polymers. 2023; 15(13):2861. https://doi.org/10.3390/polym15132861
Chicago/Turabian StylePokryshkin, Sergey, Yuliya Sypalova, Artem Ivahnov, and Aleksandr Kozhevnikov. 2023. "Optimization of Approaches to Analysis of Lignin by Thermal Decomposition" Polymers 15, no. 13: 2861. https://doi.org/10.3390/polym15132861