
Pyrolysis of Chilean Southern Lignocellulosic Biomasses: Isoconversional Kinetics Analysis and Pyrolytic Products Distribution


Abstract
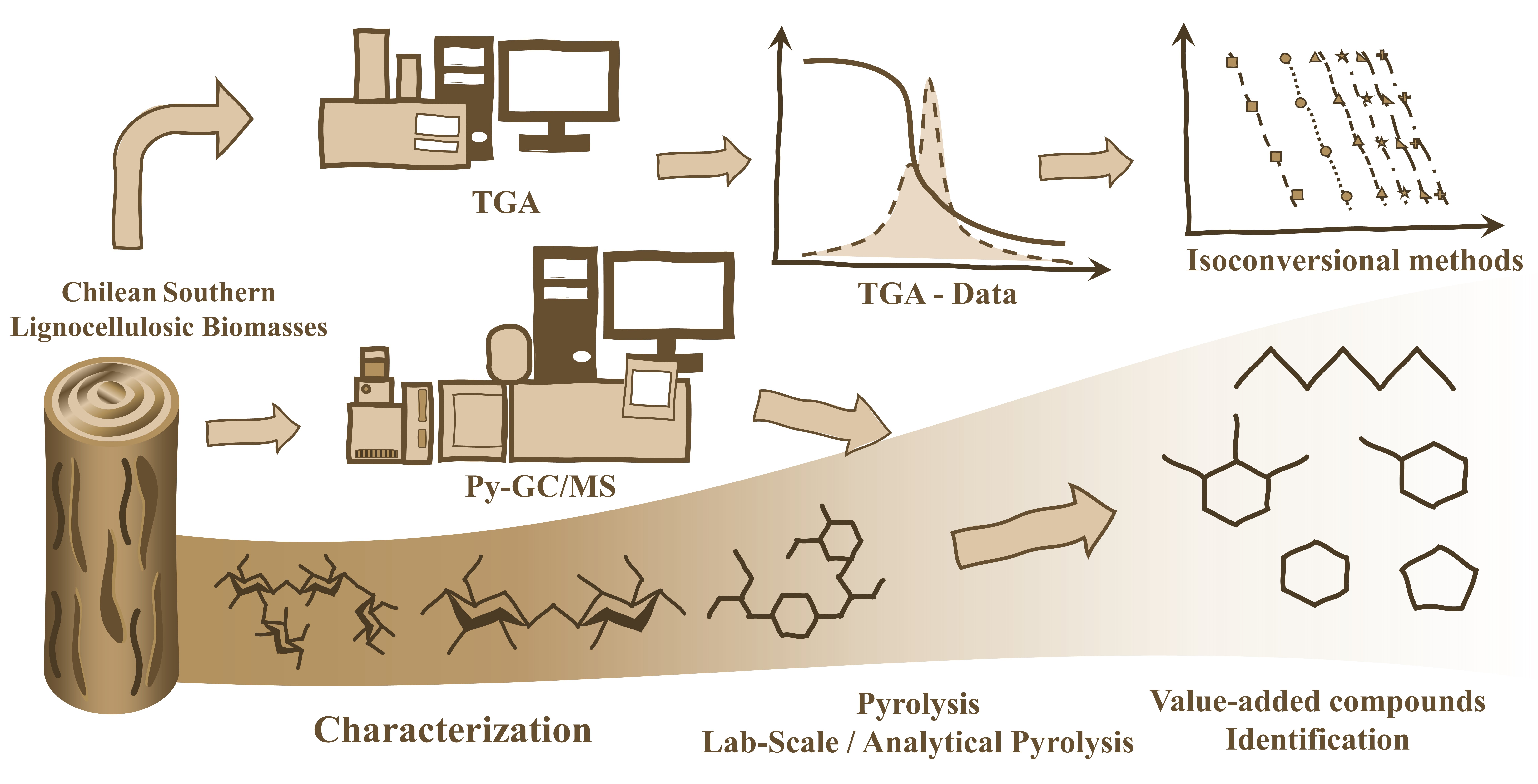
:
1. Introduction
2. Materials and Methods
2.1. Samples Preparation
2.2. Proximate Analysis
2.3. Gross Calorific Value (GCV)
2.4. Chemical Analysis
2.5. Thermogravimetric Analysis (TGA)
2.6. Processing Thermogravimetric Data
2.7. Kinetic Analysis
2.7.1. Kissinger Method
2.7.2. Kissinger–Akahira–Sunose (KAS) Method
2.7.3. Flynn–Wall–Ozawa (FWO) Method
2.7.4. Friedmann Method
2.8. Master Plots and Compensation Effect
2.9. Pyrolysis Tests
2.10. Characterisation of Bio-Oils by GC/MS
2.11. Characterisation of Permanent Gases by GC-TCD
2.12. Analytical Pyrolysis Tests (Py-GC/MS)
3. Results and Discussion
3.1. Characterisation of Biomass Samples
3.2. Effect of the Heating Rate
3.3. Kinetic Analysis
3.4. Activation Energy Evaluation Using Isoconversional Methods
3.5. Master Plot
3.6. Compensation Effect
3.7. Distribution of Pyrolysis Products
3.7.1. Chemical Composition of the Liquid Phase of Pyrolysis
3.7.2. Evolution of Gaseous Species from Pyrolysis
3.7.3. Analytical Pyrolysis
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Manikandan, S.; Subbaiya, R.; Biruntha, M.; Krishnan, R.Y.; Muthusamy, G.; Karmegam, N. Recent Development Patterns, Utilization and Prospective of Biofuel Production: Emerging Nanotechnological Intervention for Environmental Sustainability—A Review. Fuel 2022, 314, 122757. [Google Scholar] [CrossRef]
- Abbas-Abadi, M.S.; Van Geem, K.M.; Fathi, M.; Bazgir, H.; Ghadiri, M. The Pyrolysis of Oak with Polyethylene, Polypropylene and Polystyrene Using Fixed Bed and Stirred Reactors and TGA Instrument. Energy 2021, 232, 121085. [Google Scholar] [CrossRef]
- Mohan, I.; Panda, A.K.; Volli, V.; Kumar, S. An Insight on Upgrading of Biomass Pyrolysis Products and Utilization: Current Status and Future Prospect of Biomass in India. Biomass Convers. Biorefinery 2022, 1, 1–19. [Google Scholar] [CrossRef]
- Drew, D.M.; Downes, G.M.; Seifert, T.; Eckes-Shepard, A.; Achim, A. A Review of Progress and Applications in Wood Quality Modelling. Curr. For. Rep. 2022, 8, 317–332. [Google Scholar] [CrossRef]
- Azahar, A.A.; Nurhafizah, M.D.; Abdullah, N.; Ul-Hamid, A. A Review on the Palm Oil Waste Thermal Degradation Analysis and Its Kinetic Triplet Study. BioEnergy Res. 2023, 1, 1–26. [Google Scholar] [CrossRef]
- Postawa, K.; Fałtynowicz, H.; Sczygieł, J.; Beran, E.; Kułażski, M. Analysing the Kinetics of Waste Plant Biomass Pyrolysis via Thermogravimetry Modeling and Semi-Statistical Methods. Bioresour. Technol. 2022, 344, 126181. [Google Scholar] [CrossRef] [PubMed]
- Zhao, H.; Yan, H.; Liu, M.; Zhang, C.; Qin, S. Pyrolytic Characteristics and Kinetics of the Marine Green Tide Macroalgae, Enteromorpha Prolifera. Chin. J. Oceanol. Limnol. 2011, 29, 996–1001. [Google Scholar] [CrossRef]
- Park, H.J.; Park, Y.K.; Dong, J.I.; Kim, J.S.; Jeon, J.K.; Kim, S.S.; Kim, J.; Song, B.; Park, J.; Lee, K.J. Pyrolysis Characteristics of Oriental White Oak: Kinetic Study and Fast Pyrolysis in a Fluidized Bed with an Improved Reaction System. Fuel Process. Technol. 2009, 90, 186–195. [Google Scholar] [CrossRef]
- Chen, D.; Cen, K.; Zhuang, X.; Gan, Z.; Zhou, J.; Zhang, Y.; Zhang, H. Insight into Biomass Pyrolysis Mechanism Based on Cellulose, Hemicellulose, and Lignin: Evolution of Volatiles and Kinetics, Elucidation of Reaction Pathways, and Characterization of Gas, Biochar and Bio-oil. Combust. Flame 2022, 242, 112142. [Google Scholar] [CrossRef]
- Tapasvi, D.; Khalil, R.; Várhegyi, G.; Tran, K.Q.; Grønli, M.; Skreiberg, Ø. Thermal Decomposition Kinetics of Woods with an Emphasis on Torrefaction. Energy Fuels 2013, 27, 6134–6145. [Google Scholar] [CrossRef] [Green Version]
- Morales Olmos, V. Forestry and the Forest Products Sector: Production, Income and Employment, and International Trade. For. Policy Econ. 2022, 135, 102648. [Google Scholar] [CrossRef]
- Vyazovkin, S.; Achilias, D.; Fernandez-Francos, X.; Galukhin, A.; Sbirrazzuoli, N. ICTAC Kinetics Committee Recommendations for Analysis of Thermal Polymerization Kinetics. Thermochim. Acta 2022, 714, 179243. [Google Scholar] [CrossRef]
- Kissinger, H.E. Reaction Kinetics in Differential Thermal Analysis. Anal. Chem. 1957, 29, 1702–1706. [Google Scholar] [CrossRef]
- Akahira, T. Trans. Joint Convention of Four Electrical Institutes. Res. Rep. Chiba Inst. Technol. 1971, 16, 22–31. [Google Scholar]
- Flynn, J.H.; Wall, L.A. A Quick, Direct Method for the Determination of Activation Energy from Thermogravimetric Data. J. Polym. Sci. Part B Polym. Lett. 1966, 4, 323–328. [Google Scholar] [CrossRef]
- Ozawa, T. A New Method of Analysing Thermogravimetric Data. Bull. Chem. Soc. Jpn. 2006, 38, 1881–1886. [Google Scholar] [CrossRef] [Green Version]
- Friedman, H.L. Kinetics of Thermal Degradation of Char-Forming Plastics from Thermogravimetry. Application to a Phenolic Plastic. J. Polym. Sci. Part C Polym. Symp. 1964, 6, 183–195. [Google Scholar] [CrossRef]
- Starink, M.J. The Determination of Activation Energy from Linear Heating Rate Experiments: A Comparison of the Accuracy of Isoconversion Methods. Thermochim. Acta 2003, 404, 163–176. [Google Scholar] [CrossRef] [Green Version]
- Koga, N.; Vyazovkin, S.; Burnham, A.K.; Favergeon, L.; Muravyev, N.V.; Pérez-Maqueda, L.A.; Saggese, C.; Sánchez-Jiménez, P.E. ICTAC Kinetics Committee Recommendations for Analysis of Thermal Decomposition Kinetics. Thermochim. Acta 2023, 719, 179384. [Google Scholar] [CrossRef]
- Vyazovkin, S. Determining Preexponential Factor in Model-Free Kinetic Methods: How and Why? Molecules 2021, 26, 3077. [Google Scholar] [CrossRef]
- American Society for Testing and Materials. Standard Test Method for Compositional Analysis by Thermogravimetry; ASTM International: West Conshohocken, PA, USA, 2014; pp. 12959–19428. [Google Scholar]
- Savitzky, A.; Golay, M.J.E. Smoothing and Differentiation of Data by Simplified Least Squares Procedures. Anal. Chem. 1964, 36, 1627–1639. [Google Scholar] [CrossRef]
- Murray, P. Kinetics of Clay Dehydration. Clay Miner. 1955, 2, 255–264. [Google Scholar] [CrossRef] [Green Version]
- Doyle, C.D. Kinetic Analysis of Thermogravimetric Data. J. Appl. Polym. Sci. 1961, 5, 285–292. [Google Scholar] [CrossRef]
- Vyazovkin, S.; Burnham, A.K.; Criado, J.M.; Pérez-Maqueda, L.A.; Popescu, C.; Sbirrazzuoli, N. ICTAC Kinetics Committee Recommendations for Performing Kinetic Computations on Thermal Analysis Data. Thermochim. Acta 2011, 520, 1–19. [Google Scholar] [CrossRef]
- Vyazovkin, S.; Clawson, J.S.; Wight, C.A. Thermal Dissociation Kinetics of Solid and Liquid Ammonium Nitrate. Chem. Mater. 2001, 13, 960–966. [Google Scholar] [CrossRef]
- Niu, Q.; Ghysels, S.; Wu, N.; Rousseau, D.P.L.; Pieters, J.; Prins, W.; Ronsse, F. Effects of Demineralization on the Composition of Microalgae Pyrolysis Volatiles in Py-GC–MS. Energy Convers. Manag. 2022, 251, 114979. [Google Scholar] [CrossRef]
- Sanchez-Salvador, J.L.; Campano, C.; Balea, A.; Tarrés, Q.; Delgado-Aguilar, M.; Mutjé, P.; Blanco, A.; Negro, C. Critical Comparison of the Properties of Cellulose Nanofibers Produced from Softwood and Hardwood through Enzymatic, Chemical and Mechanical Processes. Int. J. Biol. Macromol. 2022, 205, 220–230. [Google Scholar] [CrossRef] [PubMed]
- Santos, T.M.; Rigual, V.; Domínguez, J.C.; Alonso, M.V.; Oliet, M.; Rodriguez, F. Fractionation of Pinus Radiata by Ethanol-Based Organosolv Process. Biomass Convers. Biorefinery 2022, 1, 1–14. [Google Scholar] [CrossRef]
- Yang, H.; Yan, R.; Chen, H.; Lee, D.H.; Zheng, C. Characteristics of Hemicellulose, Cellulose and Lignin Pyrolysis. Fuel 2007, 86, 1781–1788. [Google Scholar] [CrossRef]
- Shebani, A.N.; van Reenen, A.J.; Meincken, M. The Effect of Wood Extractives on the Thermal Stability of Different Wood Species. Thermochim. Acta 2008, 471, 43–50. [Google Scholar] [CrossRef]
- Sebio-Puñal, T.; Naya, S.; López-Beceiro, J.; Tarrío-Saavedra, J.; Artiaga, R. Thermogravimetric Analysis of Wood, Holocellulose, and Lignin from Five Wood Species. J. Therm. Anal. Calorim. 2012, 109, 1163–1167. [Google Scholar] [CrossRef]
- Kabakcı, S.B. Pyrolysis and Combustion Characteristics and Kinetics of Wood Sawdust and Wood Sawdust Hydrochar. Environ. Prog. Sustain. Energy 2020, 39, e13315. [Google Scholar] [CrossRef]
- Soria-Verdugo, A.; Morgano, M.T.; Mätzing, H.; Goos, E.; Leibold, H.; Merz, D.; Riedel, U.; Stapf, D. Comparison of Wood Pyrolysis Kinetic Data Derived from Thermogravimetric Experiments by Model-Fitting and Model-Free Methods. Energy Convers. Manag. 2020, 212, 112818. [Google Scholar] [CrossRef]
- Rollag, S.A.; Jeong, K.; Peterson, C.A.; Kim, K.H.; Brown, R.C. An Experimental and Modeling Study on the Catalytic Effects of Select Metals on the Fast Pyrolysis of Hardwood and Softwood Lignin. Green Chem. 2022, 24, 6189–6199. [Google Scholar] [CrossRef]
- Aboulkas, A.; El Harfi, K.; El Bouadili, A. Thermal Degradation Behaviors of Polyethylene and Polypropylene. Part I: Pyrolysis Kinetics and Mechanisms. Energy Convers. Manag. 2010, 51, 1363–1369. [Google Scholar] [CrossRef]
- Mishra, G.; Bhaskar, T. Insights into the Decomposition Kinetics of Groundnut Shell: An Advanced Isoconversional Approach. Renew. Energy 2022, 196, 1–14. [Google Scholar] [CrossRef]
- Li, J.; Dou, B.; Zhang, H.; Zhang, H.; Chen, H.; Xu, Y.; Wu, C. Pyrolysis Characteristics and Non-Isothermal Kinetics of Waste Wood Biomass. Energy 2021, 226, 120358. [Google Scholar] [CrossRef]
- Mampel, K.L. Zeitumsatzformeln Für Heterogene Reaktionen an Phasengrenzen Fester Körper: 1. Die Entwicklung Der Mathematischen Methode Und Die Herleitung von Flächenumsatzformeln. Z. Für Phys. Chem. 1940, 187A, 43–57. [Google Scholar] [CrossRef]
- Ahmad, M.S.; Klemeš, J.J.; Alhumade, H.; Elkamel, A.; Mahmood, A.; Shen, B.; Ibrahim, M.; Mukhtar, A.; Saqib, S.; Asif, S.; et al. Thermo-Kinetic Study to Elucidate the Bioenergy Potential of Maple Leaf Waste (MLW) by Pyrolysis, TGA and Kinetic Modelling. Fuel 2021, 293, 120349. [Google Scholar] [CrossRef]
- Lu, Q.; Yang, X.C.; Dong, C.Q.; Zhang, Z.F.; Zhang, X.M.; Zhu, X.F. Influence of Pyrolysis Temperature and Time on the Cellulose Fast Pyrolysis Products: Analytical Py-GC/MS Study. J. Anal. Appl. Pyrolysis 2011, 92, 430–438. [Google Scholar] [CrossRef]
- Huang, Y.; Wang, L.; Chao, Y.; Nawawi, D.S.; Akiyama, T.; Yokoyama, T.; Matsumoto, Y. Analysis of Lignin Aromatic Structure in Wood Based on the IR Spectrum. J. Wood Chem. Technol. 2012, 32, 294–303. [Google Scholar] [CrossRef]
- Lopes, F.J.F.; Silvério, F.O.; Baffa, D.C.F.; Loureiro, M.E.; Barbosa, M.H.P. Determination of Sugarcane Bagasse Lignin S/G/H Ratio by Pyrolysis GC/MS. J. Wood Chem. Technol. 2011, 31, 309–323. [Google Scholar] [CrossRef]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | Volatile Matter (wt.%) | Fixed Carbon (wt.%) | Ash (wt.%) | Moisture (wt.%) | GCV a (MJ·kg−1) | GCV b (MJ·kg−1) | GCV c (MJ·kg−1) |
|---|---|---|---|---|---|---|---|
| PR (Pinus radiata) | 83.41 | 13.94 | 2.65 | 9.48 | 17.82 | 20.98 | 32.41 |
| NO (Nothofagus obliqua) | 85.74 | 12.62 | 3.30 | 8.12 | 17.89 | 20.72 | 24.93 |
| EN (Eucalyptus nitens) | 85.14 | 13.34 | 1.51 | 5.86 | 17.59 | 18.26 | 29.33 |
| EC (Eucryphia cordifolia) | 81.97 | 15.18 | 2.87 | 9.09 | 17.52 | 19.06 | 27.83 |
| ND (Nothofagus domberyi) | 82.53 | 16.08 | 1.40 | 11.38 | 16.04 | 18.85 | 24.44 |
| Sample | Holocellulose (%) | Cellulose (%) | Extractives (%) | Lignin (%) | Hemicellulose (%) a |
|---|---|---|---|---|---|
| PR | 70.08 | 34.25 | 1.79 | 28.12 | 35.83 |
| NO | 70.93 | 35.38 | 2.43 | 27.10 | 35.55 |
| EN | 73.76 | 32.30 | 2.52 | 24.49 | 41.46 |
| EC | 73.01 | 34.14 | 1.86 | 25.03 | 38.87 |
| ND | 72.82 | 36.13 | 1.45 | 25.39 | 36.69 |
| Sample | Heating Rate (°C·min−1) | Tonset (°C) | T1 (°C) | Tm (°C) | αm | DTGm (mg·s−1) | Toffset (°C) | αoffset | %R |
|---|---|---|---|---|---|---|---|---|---|
| PR | 5 | 192 | 311 | 348 | 0.63 | 0.0041 | 386 | 0.83 | 13.9 |
| 10 | 198 | 325 | 359 | 0.66 | 0.0085 | 403 | 0.85 | 14.5 | |
| 20 | 213 | 341 | 380 | 0.66 | 0.0158 | 426 | 0.86 | 10.7 | |
| 40 | 225 | 359 | 398 | 0.68 | 0.0324 | 452 | 0.89 | 14.2 | |
| NO | 5 | 198 | 275 | 348 | 0.66 | 0.0053 | 384 | 0.84 | 8.8 |
| 10 | 200 | 286 | 359 | 0.68 | 0.0090 | 398 | 0.86 | 10.1 | |
| 20 | 203 | 307 | 379 | 0.70 | 0.0179 | 434 | 0.89 | 13.7 | |
| 40 | 205 | 328 | 400 | 0.72 | 0.0329 | 448 | 0.90 | 14.7 | |
| EN | 5 | 196 | 279 | 336 | 0.59 | 0.0055 | 386 | 0.82 | 7.6 |
| 10 | 199 | 291 | 343 | 0.60 | 0.0105 | 395 | 0.88 | 10.7 | |
| 20 | 202 | 304 | 364 | 0.62 | 0.0189 | 428 | 0.88 | 10.7 | |
| 40 | 234 | 320 | 381 | 0.72 | 0.0372 | 438 | 0.91 | 12.8 | |
| EC | 5 | 188 | 286 | 349 | 0.64 | 0.0056 | 383 | 0.82 | 11.1 |
| 10 | 192 | 297 | 357 | 0.64 | 0.0092 | 405 | 0.86 | 13.1 | |
| 20 | 205 | 309 | 380 | 0.66 | 0.0167 | 429 | 0.86 | 12.1 | |
| 40 | 219 | 327 | 398 | 0.66 | 0.0354 | 446 | 0.87 | 13.6 | |
| ND | 5 | 172 | 276 | 339 | 0.62 | 0.0046 | 374 | 0.80 | 13.5 |
| 10 | 172 | 283 | 345 | 0.62 | 0.0080 | 394 | 0.81 | 17.3 | |
| 20 | 175 | 302 | 364 | 0.64 | 0.0164 | 417 | 0.85 | 16.4 | |
| 40 | 198 | 328 | 376 | 0.60 | 0.0346 | 436 | 0.85 | 15.0 |
| Sample | Activation Energies, Ea () | ||
|---|---|---|---|
| KAS Method | FWO Method | FR Method | |
| PR | 139.25 | 142.07 | 140.70 |
| NO | 131.60 | 134.70 | 140.92 |
| EN | 137.34 | 140.10 | 148.45 |
| EC | 133.50 | 136.56 | 138.38 |
| ND | 133.49 | 144.91 | 158.08 |
| α | PR | NO | EN | EC | ND | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Eα (kJ·mol−1) | Aα (min−1) | R2 | Eα (kJ·mol−1) | Aα (min−1) | R2 | Eα (kJ·mol−1) | Aα (min−1) | R2 | Eα (kJ·mol−1) | Aα (min−1) | R2 | Eα (kJ·mol−1) | Aα (min−1) | R2 | |
| 0.05 | 148.76 | 5.83 × 1012 | 0.962 | 115.38 | 1.20 × 1010 | 0.982 | 124.85 | 9.85 × 1010 | 0.976 | 122.02 | 3.33 × 1010 | 0.957 | 125.12 | 7.42 × 1010 | 0.978 |
| 0.10 | 141.77 | 1.58 × 1012 | 0.982 | 123.96 | 6.01 × 1010 | 0.983 | 128.70 | 2.05 × 1011 | 0.986 | 130.10 | 1.49 × 1011 | 0.981 | 133.87 | 3.80 × 1011 | 0.981 |
| 0.15 | 137.46 | 7.04 × 1011 | 0.985 | 128.33 | 1.37 × 1011 | 0.988 | 135.45 | 7.37 × 1011 | 0.981 | 135.16 | 3.80 × 1011 | 0.980 | 140.51 | 1.31 × 1012 | 0.983 |
| 0.20 | 138.27 | 8.20 × 1011 | 0.986 | 131.46 | 2.45 × 1011 | 0.989 | 142.70 | 2.92 × 1012 | 0.979 | 137.70 | 6.09 × 1011 | 0.978 | 147.59 | 4.94 × 1012 | 0.980 |
| 0.25 | 139.87 | 1.11 × 1012 | 0.983 | 139.09 | 1.02 × 1012 | 0.989 | 150.36 | 1.25 × 1013 | 0.974 | 138.45 | 7.01 × 1011 | 0.975 | 153.86 | 1.59 × 1013 | 0.983 |
| 0.30 | 141.75 | 1.57 × 1012 | 0.985 | 144.08 | 2.61 × 1012 | 0.988 | 149.33 | 1.03 × 1013 | 0.976 | 141.51 | 1.24 × 1012 | 0.973 | 158.82 | 4.02 × 1013 | 0.983 |
| 0.35 | 139.87 | 1.11 × 1012 | 0.983 | 147.93 | 5.38 × 1011 | 0.990 | 148.30 | 8.47 × 1012 | 0.978 | 140.36 | 9.99 × 1011 | 0.974 | 159.76 | 4.79 × 1013 | 0.980 |
| 0.40 | 138.06 | 7.88 × 1011 | 0.986 | 147.79 | 5.23 × 1012 | 0.987 | 149.10 | 9.86 × 1012 | 0.977 | 138.31 | 6.83 × 1011 | 0.983 | 161.68 | 6.86 × 1013 | 0.984 |
| 0.45 | 137.83 | 7.54 × 1011 | 0.985 | 146.02 | 3.75 × 1012 | 0.986 | 151.01 | 1.42 × 1013 | 0.980 | 135.45 | 4.02 × 1011 | 0.986 | 161.13 | 6.19 × 1013 | 0.989 |
| 0.50 | 137.54 | 7.14 × 1011 | 0.986 | 145.43 | 3.36 × 1012 | 0.989 | 151.12 | 1.45 × 1013 | 0.984 | 135.05 | 3.73 × 1011 | 0.986 | 159.94 | 4.96 × 1013 | 0.989 |
| 0.55 | 137.99 | 7.78 × 1011 | 0.988 | 144.31 | 2.73 × 1012 | 0.992 | 152.14 | 1.75 × 1013 | 0.983 | 136.21 | 4.62 × 1011 | 0.986 | 159.73 | 4.77 × 1013 | 0.991 |
| 0.60 | 138.14 | 7.99 × 1011 | 0.992 | 142.97 | 2.12 × 1012 | 0.992 | 153.21 | 2.15 × 1013 | 0.983 | 140.88 | 1.10 × 1012 | 0.986 | 167.21 | 1.93 × 1014 | 0.991 |
| 0.65 | 139.12 | 9.61 × 1011 | 0.992 | 147.31 | 4.78 × 1012 | 0.994 | 154.66 | 2.83 × 1013 | 0.990 | 144.33 | 2.09 × 1012 | 0.985 | 169.43 | 2.92 × 1014 | 0.990 |
| 0.70 | 146.79 | 4.03 × 1012 | 0.999 | 150.78 | 9.17 × 1012 | 0.993 | 161.97 | 1.13 × 1014 | 0.976 | 147.23 | 3.57 × 1012 | 0.983 | 177.79 | 1.39 × 1015 | 0.989 |
| 0.75 | 147.34 | 4.47 × 1012 | 0.993 | 158.98 | 4.26 × 1013 | 0.989 | 173.92 | 1.10 × 1015 | 0.957 | 152.88 | 1.02 × 1013 | 0.982 | 194.81 | 3.35 × 1016 | 0.982 |
| Avg | 140.70 | 1.73 × 1012 | 140.92 | 5.54 × 1012 | 148.45 | 9.03 × 1013 | 138.38 | 1.53 × 1012 | 158.08 | 2.38 × 1015 | |||||
| Sample | Biochar (wt. (%)) | Liquid (wt. (%)) | Gas (wt. (%)) * |
|---|---|---|---|
| PR | 33.95 | 48.06 | 17.99 |
| NO | 34.04 | 37.37 | 28.59 |
| EN | 35.78 | 38.83 | 25.39 |
| EC | 33.00 | 28.85 | 38.15 |
| ND | 31.11 | 41.93 | 26.96 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cerda-Barrera, C.; Fernández-Andrade, K.J.; Alejandro-Martín, S. Pyrolysis of Chilean Southern Lignocellulosic Biomasses: Isoconversional Kinetics Analysis and Pyrolytic Products Distribution. Polymers 2023, 15, 2698. https://doi.org/10.3390/polym15122698
Cerda-Barrera C, Fernández-Andrade KJ, Alejandro-Martín S. Pyrolysis of Chilean Southern Lignocellulosic Biomasses: Isoconversional Kinetics Analysis and Pyrolytic Products Distribution. Polymers. 2023; 15(12):2698. https://doi.org/10.3390/polym15122698
Chicago/Turabian StyleCerda-Barrera, Cristian, Kevin J. Fernández-Andrade, and Serguei Alejandro-Martín. 2023. "Pyrolysis of Chilean Southern Lignocellulosic Biomasses: Isoconversional Kinetics Analysis and Pyrolytic Products Distribution" Polymers 15, no. 12: 2698. https://doi.org/10.3390/polym15122698