1. Introduction
Self-organization of amphiphilic polymers in aqueous solution is of high potential importance in a variety of applications such as paints, coatings, drug delivery, electronics, agriculture and personal goods. The conformational behavior of polymer chains in different regional environments can be explained using the Flory–Huggins theory. Flory [
1] describes conformations of uncharged polymers in terms of the theta condition, which could be a theta solvent or theta temperature. When a polymer chain is in the theta condition, it behaves as an ideal chain where polymer–polymer interactions are balanced with polymer–solvent interactions, and the radius of gyration is equal to the random walk configuration. Any deviation from the theta condition can cause the radius Rg to change either due to swelling in a good solvent, greater than theta, or collapse in a poor solvent, less than theta. However, these conformational changes are more complex for charged polyelectrolytes in solution [
2,
3,
4,
5].
Colloidal unimolecular polymers or CUPs are nanoscale, charge-stabilized, single-chain nanoparticles made from a single polymer chain and have a well-balanced number of hydrophobic and hydrophilic units [
6]. The hydrophilic units can be anionic or cationic. The polymer chain is collapsed into a particle through a simple process called water reduction.
Figure 1 shows the schematics of the water-reduction process to form CUP particles. The water-reduction process begins with the dissolving of the polymer in a low-boiling, water-miscible solvent. The boiling point of the solvent should be less than that of water since the solvent will be stripped off at the end of the reduction process. THF, used in this study, is a good example of a water-miscible solvent that has good solubility for many polymers, and has a low boiling point of 66 C. The next step is to form the salt or ionic group, in this case, by neutralizing the acid groups—carboxylic acids—using any base such as sodium hydroxide, triethyl amine, ammonia, etc. The base should be added slowly, preferably using a peristaltic pump. Due to the low dielectric of THF, the carboxylate anion and the sodium counterion exist as a tight or intimate ion pair. The repulsive force between the carboxylate anions on the polymer chain is negligible. The polymer chains form inter-chain and intra-chain salt association (see
Figure 2) via the sodium-carboxylate group, which causes a small rise in viscosity. The next step is to add water very slowly (1.8 g/min) with stirring to ensure homogenous composition of the solvent throughout the mixture. For water, which is a poor solvent for the polymer, any localized spikes in concentration can cause the polymer to precipitate instead of a proper unimolecular collapse. As the water is being added, the dielectric of the solvent mixture increases [
7], as seen in
Figure 3. The carboxylate anions start repelling each other more strongly over a longer distance as the dielectric of the media increase. As a result, the polymer chain will become more elongated, and the viscosity will increase as more water is added. This trend will continue until the concentration of water in the solvent mixture reaches a point where it becomes a poor solvent for the polymer and the chain collapses into a spheroidal particle. Here, the transition from coil to globule is triggered by changing the dielectric and solubility parameters of the solvent. The changes in the thermodynamic quality of the solvent makes the polymer–polymer interactions stronger than polymer–solvent interactions, which causes the chain to collapse into a globule. The collapse of the chain is such that the hydrophobic segments form the interior of the particle, and the charged groups are on the surface, as shown in
Figure 4. The self-organization of polymer chains into CUPs is similar to that of micelle formation in surfactants. CUPs have a lot of utility in the field of coatings due to their zero-VOC (volatile organic content), low cost and easy synthesis. They have been used as a resin that can be cured with melamine or aziridine [
8,
9], an additive for the freeze–thaw stability and wet-edge retention [
10] of latex paints or as a catalyst [
11]. They can also be potential drug-delivery systems. CUPs have been extremely useful in studying the properties of bound or surface water [
12,
13], and in understanding water evaporation behavior [
14], the electroviscous effect [
15] and surface tension [
16].
A polyelectrolyte or a polymer chain containing several ionic groups can form many different conformations depending on the charge density on the chain and its solvent environment. The conformations of polyelectrolytes have been modeled using an electrostatic blob and scaling theory, which was first developed by DeGennes and Pfeuty and later reviewed by Dobrynin [
17]. According to the theory, a neutral polymer in a poor solvent such as water will collapse into a spheroidal globule. When charges are present on the chain, it collapses into an electrostatic blob, dumbbell or a pearl necklace, depending on the fraction of charges present on the chain. Kirwan [
2] observed conformational changes for polyvinyl amine in water at different pH values. At a low pH = 3, the polymer chain was highly charged and in an extended conformation. Increasing the pH transitioned the chain into a pearl necklace structure. Above pH 9, the polymer collapsed to a globule due to attractive hydrophobic interactions between polymer segments in a poor solvent condition. Similar observations were made by DeMelo [
3] using polyacrylic acid by going from high pH to low. The conformational behavior in polymers allows the synthesis of polymers capable of forming a single-chain nanoparticle [
18,
19,
20]. The coil-to-globule transition can be triggered by changing the temperature [
21] or by changing the solvent quality such as solvent composition [
4,
22,
23], dielectric [
24], or pH [
25].
Li [
26] used hydrophobic blocks of the anticancer drug paclitaxel and grafted it onto blocks of polyether ester to produce a self-assembled multichain polymeric micelle as a drug-delivery system. When the block co-polymer was placed in an aqueous environment with adjusted pH, the hydrophilic polyether ester was oriented into the water phase, leaving the hydrophobic paclitaxel oriented toward the interior domain. Morishima reported micelle-like behavior in single-chain polyelectrolytes [
27] using a random copolymer of a 1:1 monomer ratio of hydrophilic and hydrophobic monomers. The chains were collapsed into unimolecular micelles by dissolving the polymer at very low concentration in aqueous NaOH to obtain particles of 5.5 nm in diameter. The use of a change in solvent composition to achieve a coil-to-globule transition such as CUPs was reported by Aseyev [
4]. In their work, polymethacryloyl ethyl trimethyl ammonium methyl sulfate (PMETMMS) was examined in a water/acetone mixture wherein acetone was a non-solvent. Collapse of the polymer chains occurred at a 0.80 mass fraction of acetone in aqueous solution, as observed by a decrease in the reduced viscosity, the radius of gyration and hydrodynamic radius. In an earlier report on the synthesis of CUPs [
6], viscosity was used to determine the composition of THF/water mixture required for the coil-to-globule transition of MMA–MAA copolymer. Similar to Aseyev’s observation, the viscosity of the solution dropped when the polymer chains collapsed into CUP particles. The transition occurred at roughly 60% water and 40% THF composition for the CUP example. It should be noted that, unlike other studies, in the CUP system, the good solvent is removed after the particle forms. This solvent removal secures the polymer’s spheroidal conformation and removes all VOCs.
Non-unimolecular collapse can also be observed in the case of polyurethane dispersions, which are used in the coating industry, wherein the polymer is synthesized in acetone and then followed by the addition of water. When the acetone is removed from the resin blend, the chain collapses into multi-chain aggregates/non-unimolecular particles with diameters of approximately 25 nm [
28]. For CUPs, the concentration of polymer in the solution is low enough to prevent chain overlapping or entangling, thereby ensuring that the collapse is unimolecular/single-chain. The unimolecular collapse is confirmed by measuring the particle size using dynamic light scattering (DLS) and overlapping its distribution with the particle size, calculated from the absolute molecular weight measurements using GPC. For unimolecular collapse, the measured and calculated size distribution match [
6,
20]. The addition of the non-solvent water will alter the dielectric and solubility parameters. The changing solvent composition will have an affect, which will be highly dependent upon the polymer composition in terms of ionic groups and the size and number of hydrophobic groups. The point of collapse has both charge effects and solubility considerations.
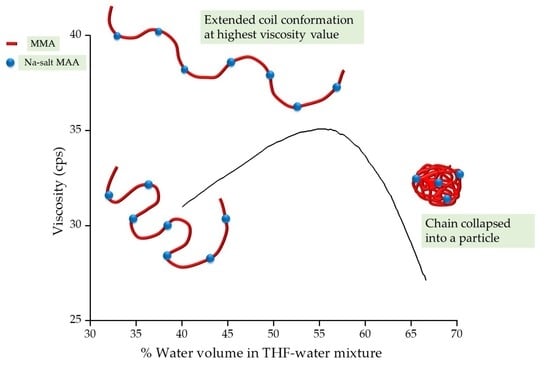
The process of water reduction leading to the collapse of the polymer chain into a particle can be tracked by measuring the viscosity as water is being added to it.
Figure 1 illustrates the conformational changes of a polymer and viscosity behavior during the water-reduction process. The composition of the water/THF mixture where the polymer chain transitions from a coil to globule is called the collapse point or collapse composition. In an earlier publication describing the synthesis of CUPs [
6], preliminary work for the determination of collapse composition was conducted using a cone-and-plate viscometer (Brookfield). The method used was a batch process wherein the water-reduction process was carried out in a separate vessel, and small aliquots of the sample were taken from that solution at regular intervals for viscosity measurement. As the measured sample was not returned to the solution, the concentration had to be corrected every time. Additionally, due to the high volatility of THF, it was difficult to prevent evaporation loss despite the enclosure provided by the instrument. The amount of sample required to measure the viscosity on the instrument was very small (0.5 mL). Hence, even a small loss in THF would significantly change the composition of the solvent.
Due to the complexity, tediousness and high error margin of the cone-and-plate system, a new protocol was developed to measure viscosity continuously during the reduction using a different type of viscometer called the vibration viscometer. The set-up developed in this study allowed, for the first time, rapid and precise determination of the collapse point where the polymer chain transforms into a collapsed particle. This further enabled us to study the effects of polymer chain composition on the solvent composition required for collapse. The structure of the polymer was also changed by using different amounts and sizes of hydrophobic monomers, and the type of base used for neutralizing the polymer was also investigated. The technique developed in this study can potentially be used to study Flory–Huggins collapse behavior in other polymeric systems.
















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}