
Self-Healable, Strong, and Tough Polyurethane Elastomer Enabled by Carbamate-Containing Chain Extenders Derived from Ethyl Carbonate


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Experimental
2.1. Materials
2.2. Synthesis of BDM, BDH
2.3. Synthesis of Polyurethane Elastomers Using BDM and BDH as Chain Extenders
2.4. Tensile Tests
2.5. Adhesion Tests
2.6. Characterizations
3. Results and Discussion
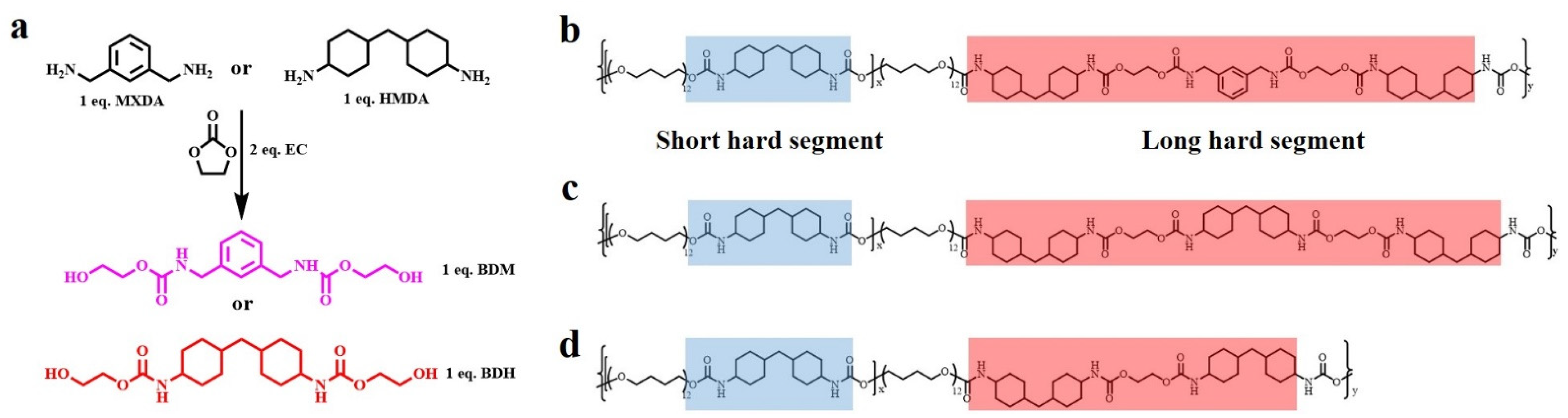
3.1. Molecular Design
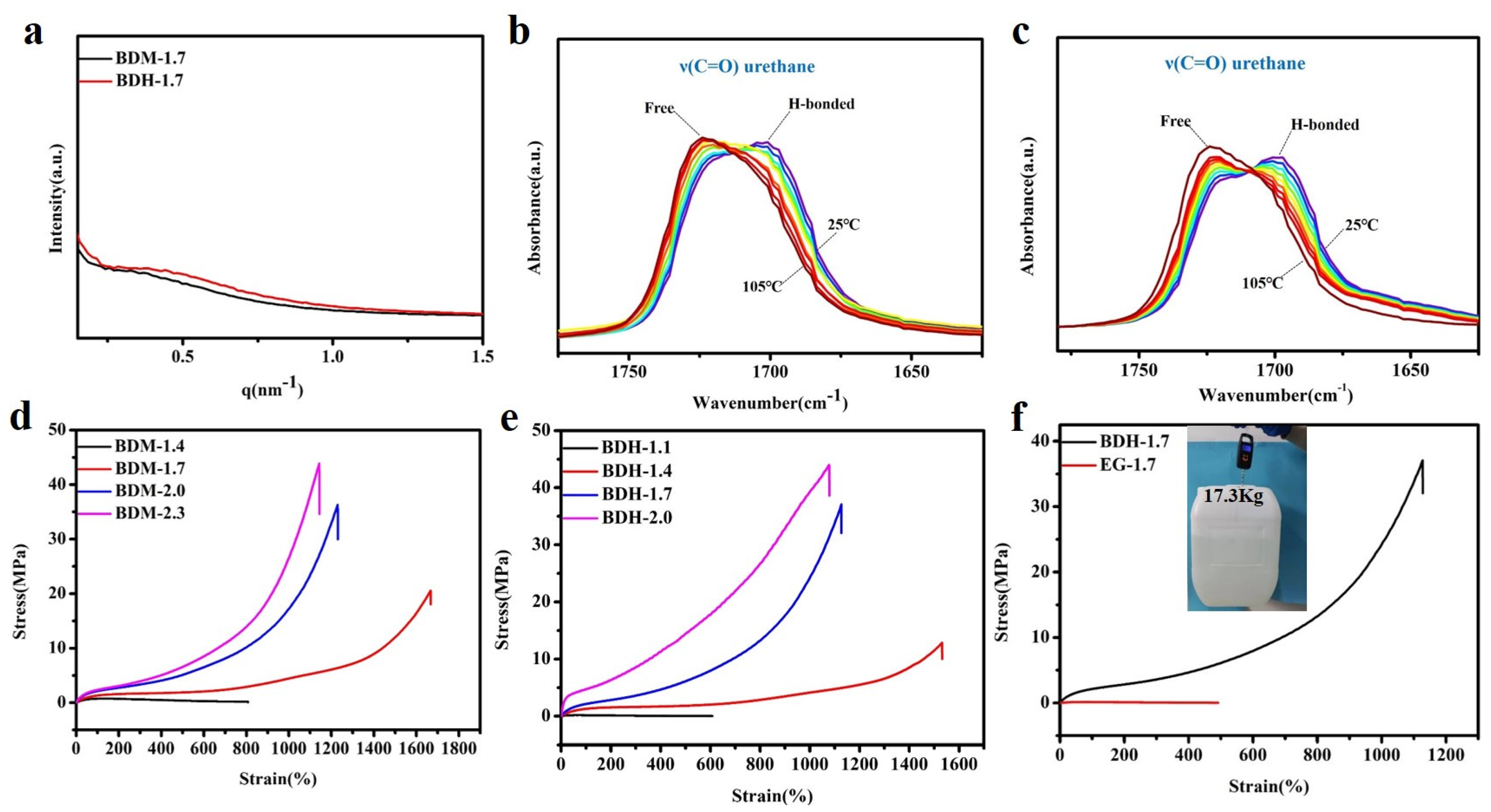
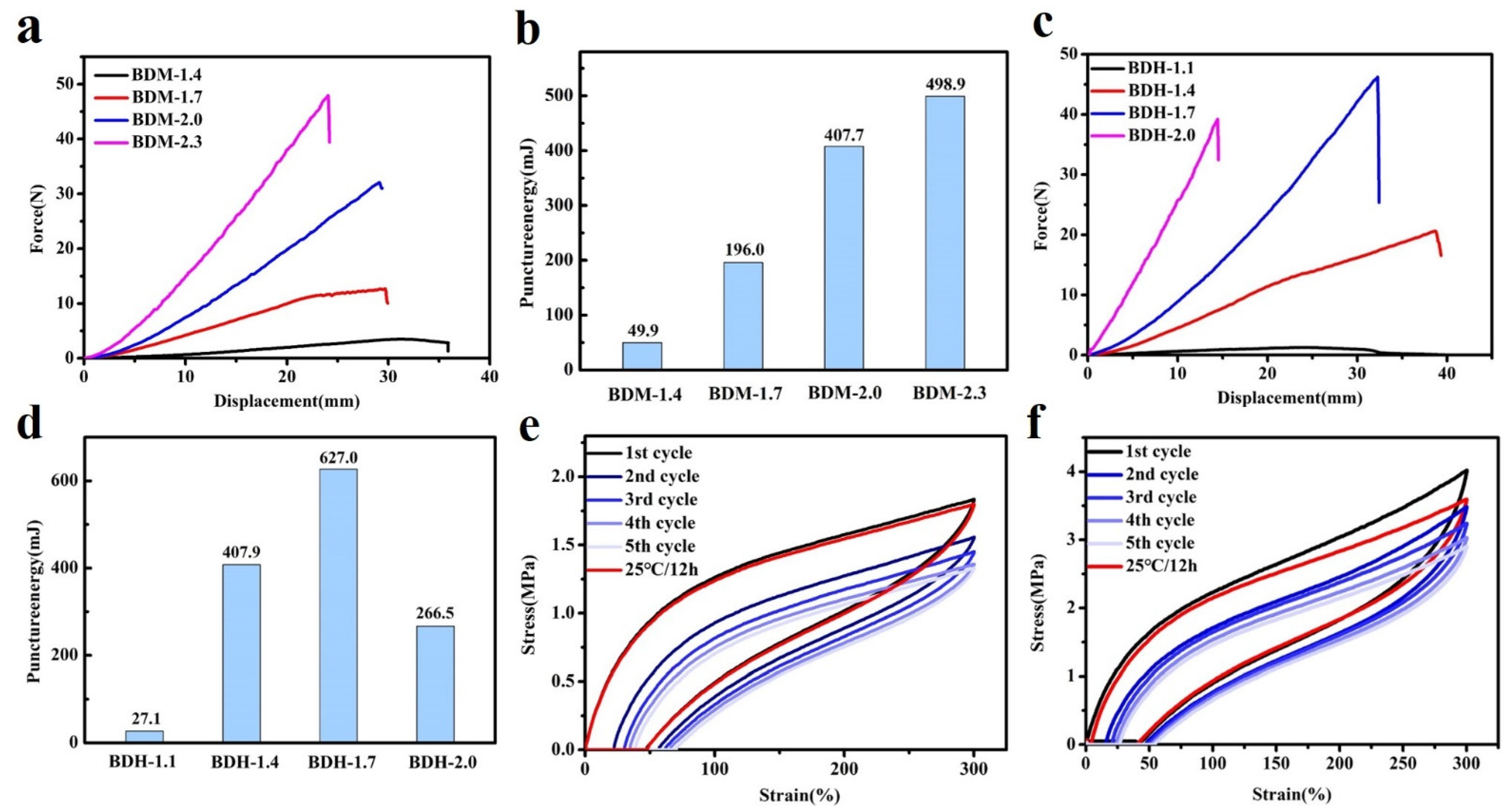
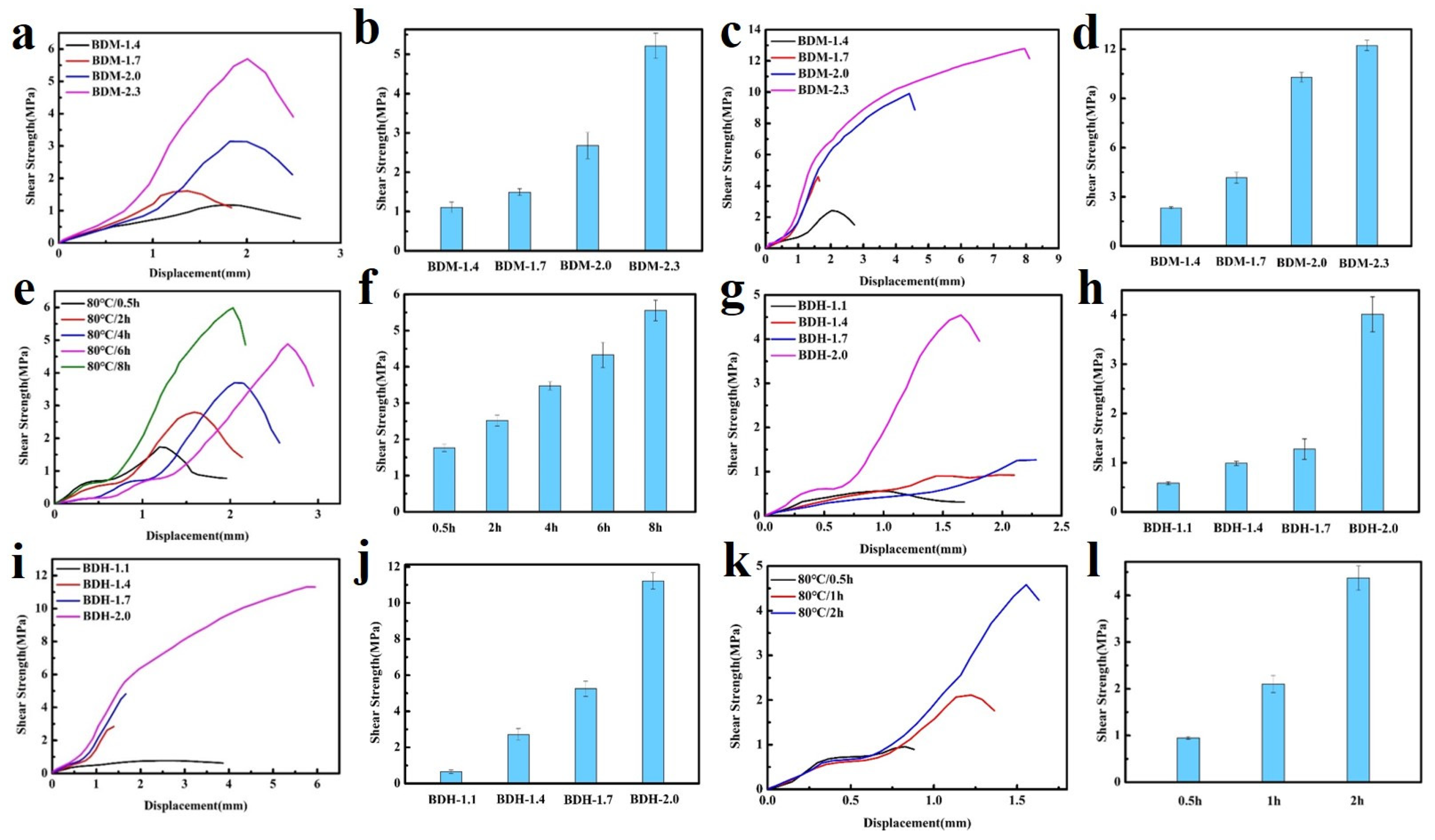
3.2. Mechanical Properties

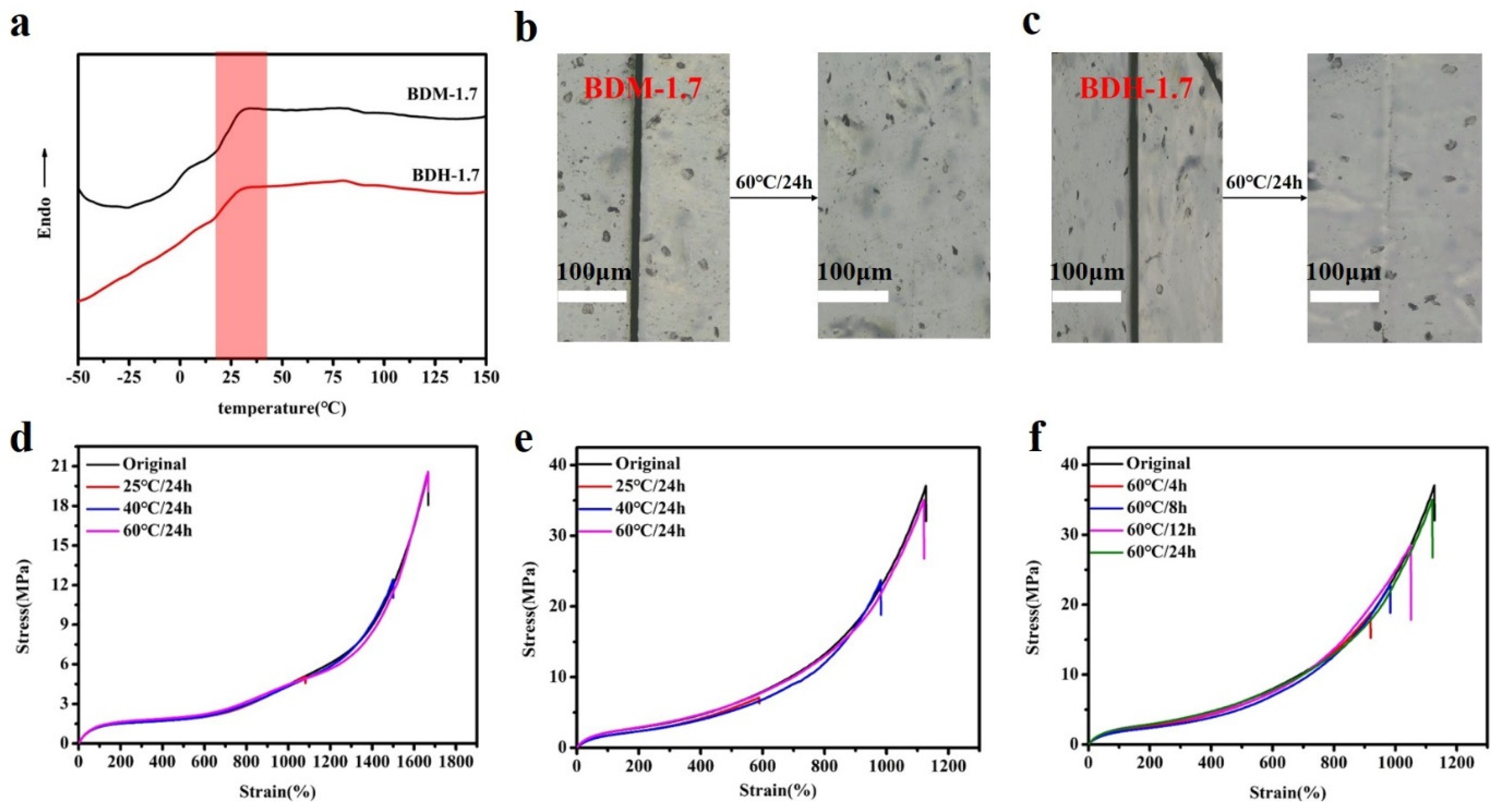
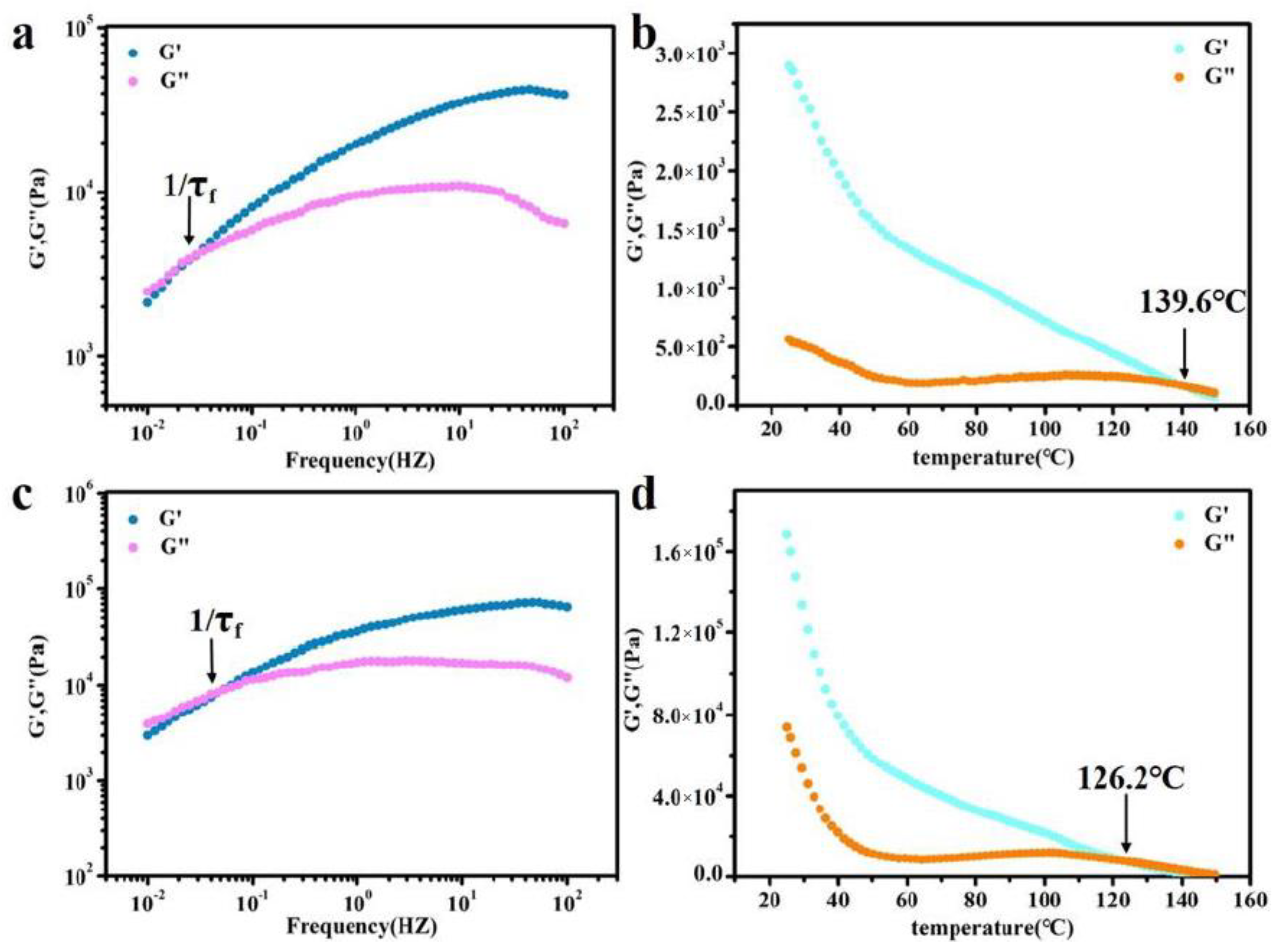
3.3. Self-Healing Properties
3.4. Adhesion Properties
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Fang, Y.; Du, X.; Du, Z.; Wang, H.; Cheng, X. Light and heat-triggered polyurethane based on dihydroxyl anthracene derivatives for self-healing applications. J. Mater. Chem. A 2017, 5, 8010–8017. [Google Scholar] [CrossRef]
- Liang, Z.; Huang, D.; Zhao, L.; Nie, Y.; Zhou, Z.; Hao, T.; Li, S. Self-healing Polyurethane Elastomer Based on Molecular Design: Combination of Reversible Hydrogen Bonds and High Segment Mobility. J. Inorg. Organomet. Polym. Mater. 2021, 31, 683–694. [Google Scholar] [CrossRef]
- Wu, H.; Xie, H.; Tian, X.; Sun, Y.; Shi, B.; Zhou, Y.; Sheng, D.; Liu, X. Hard, tough and fast self-healing thermoplastic polyurethane. Prog. Org. Coat. 2021, 159, 106409. [Google Scholar] [CrossRef]
- Chen, Y.; Kushner, A.M.; Williams, G.A.; Guan, Z. Multiphase design of autonomic self-healing thermoplastic elastomers. Nat. Chem. 2012, 4, 467–472. [Google Scholar] [CrossRef]
- Yuan, Y.; Yin, T.; Rong, M.; Zhang, M. Self healing in polymers and polymer composites. Concepts, realization and outlook: A review. Express Polym. Lett. 2008, 2, 238–250. [Google Scholar] [CrossRef]
- Kragl, M.; Knapp, D.; Nacu, E.; Khattak, S.; Maden, M.; Epperlein, H.H.; Tanaka, E.M. Cells keep a memory of their tissue origin during axolotl limb regeneration. Nature 2009, 460, 60–65. [Google Scholar] [CrossRef]
- Song, K.; Ye, W.; Gao, X.; Fang, H.; Zhang, Y.; Zhang, Q.; Li, X.; Yang, S. Synergy between dynamic covalent boronic ester and boron–nitrogen coordination: Strategy for self-healing polyurethane elastomers at room temperature with unprecedented mechanical properties. Mater. Horiz. 2021, 8, 216–223. [Google Scholar] [CrossRef]
- Zhang, W.; Wang, M.; Zhou, J.; Sheng, Y.; Xu, M.; Jiang, X.; Ma, Y.; Lu, X. Preparation of room-temperature self-hea-ling elastomers with high strength based on multiple dynamic bonds. Eur. Polym. J. 2021, 156, 110614. [Google Scholar] [CrossRef]
- Cash, J.J.; Kubo, T.; Bapat, A.P.; Sumerlin, B.S. Room-Temperature Self-Healing Polymers Based on Dynamic-Covalent Boronic Esters. Macromolecules 2015, 48, 2098–2106. [Google Scholar] [CrossRef]
- Feng, Z.; Yu, B.; Hu, J.; Zuo, H.; Li, J.; Sun, H.; Ning, N.; Tian, M. Multifunctional Vitrimer-Like Polydimethylsiloxane (PDMS): Recyclable, Self-Healable, and Water-Driven Malleable Covalent Networks Based on Dynamic Imine Bond. Ind. Eng. Chem. Res. 2019, 58, 1212–1221. [Google Scholar] [CrossRef]
- Ji, S.; Cao, W.; Yu, Y.; Xu, H. Visible-Light-Induced Self-Healing Diselenide-Containing Polyurethane Elastomer. Adv. Mater. 2015, 27, 7740–7745. [Google Scholar] [CrossRef] [PubMed]
- Kuhl, N.; Bode, S.; Bose, R.K.; Vitz, J.; Seifert, A.; Hoeppener, S.; Garcia, S.J.; Spange, S.; van der Zwaag, S.; Hager, M.D.; et al. Acylhydrazones as Reversible Covalent Crosslinkers for Self-Healing Polymers. Adv. Funct. Mater. 2015, 25, 3295–3301. [Google Scholar] [CrossRef]
- Peng, Y.; Yang, Y.; Wu, Q.; Wang, S.; Huang, G.; Wu, J. Strong and tough self-healing elastomers enabled by dual r-eversible networks formed by ionic interactions and dynamic covalent bonds. Polymer 2018, 157, 172–179. [Google Scholar] [CrossRef]
- Rekondo, A.; Martin, R.; Ruiz de Luzuriaga, A.; Cabañero, G.; Grande, H.J.; Odriozola, I. Catalyst-free room-temperature self-healing elastomers based on aromatic disulfide metathesis. Mater. Horiz. 2014, 1, 237–240. [Google Scholar] [CrossRef]
- Xu, C.; Cui, R.; Fu, L.; Lin, B. Recyclable and heat-healable epoxidized natural rubber/bentonite composites. Compos. Sci. Technol. 2018, 167, 421–430. [Google Scholar] [CrossRef]
- Chen, S.; Mo, F.; Yang, Y.; Stadler, F.J.; Chen, S.; Yang, H.; Ge, Z. Development of zwitterionic polyurethanes with multi-shape memory effects and self-healing properties. J. Mater. Chem. A 2015, 3, 2924–2933. [Google Scholar] [CrossRef]
- Li, C.H.; Zuo, J.L. Self-Healing Polymers Based on Coordination Bonds. Adv. Mater. 2020, 32, 1903762. [Google Scholar] [CrossRef]
- Song, Y.; Liu, Y.; Qi, T.; Li, G. Towards Dynamic but Supertough Healable Polymers through Biomimetic Hierarchi-cal Hydrogen-Bonding Interactions. Angew. Chem. Int. Ed. 2018, 57, 13838–13842. [Google Scholar] [CrossRef]
- Wang, Z.; An, G.; Zhu, Y.; Liu, X.; Chen, Y.; Wu, H.; Wang, Y.; Shi, X.; Mao, C. 3D-printable self-healing and mechanically reinforced hydrogels with host–guest non-covalent interactions integrated into covalently linked networks. Mater. Horiz. 2019, 6, 733–742. [Google Scholar] [CrossRef]
- An, S.Y.; Noh, S.M.; Nam, J.H.; Oh, J.K. Dual Sulfide–Disulfide Crosslinked Networks with Rapid and Room Temperature Self-Healability. Macromol. Rapid Commun. 2015, 36, 1255–1260. [Google Scholar] [CrossRef]
- Bekas, D.G.; Tsirka, K.; Baltzis, D.; Paipetis, A.S. Self-healing materials: A review of advances in materials, evaluation,characterization and monitoring techniques. Compos. Part B Eng. 2016, 87, 92–119. [Google Scholar] [CrossRef]
- Priemel, T.; Degtyar, E.; Dean, M.N.; Harrington, M.J. Rapid self-assembly of complex biomolecular architectures during musselbyssus biofabrication. Nat. Commun. 2017, 8, 14539. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Appel, A.K.; Thomann, R.; Mülhaupt, R. Hydroxyalkylation and Polyether Polyol Grafting of Graphene Tailored for Graphene/Polyurethane Nanocomposites. Macromol. Rapid Commun. 2013, 34, 1249–1255. [Google Scholar] [CrossRef] [PubMed]
- Hunley, M.T.; Pötschke, P.; Long, T.E. Melt Dispersion and Electrospinning of Non-Functionalized Multiwalled Carbo-n Nanotubes in Thermoplastic Polyurethane. Macromol. Rapid Commun. 2009, 30, 2102–2106. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Pan, Y.; Shen, C.; Liu, C.; Liu, X. Facile Thermally Impacted Water-Induced Phase Separation Approach for the Fabrication of Skin-Free Thermoplastic Polyurethane Foam and Its Recyclable Counterpart for Oil–Water Separation. Macromol. Rapid Commun. 2018, 39, 1800635. [Google Scholar] [CrossRef] [PubMed]
- Nevejans, S.; Ballard, N.; Fernández, M.; Reck, B.; Asua, J.M. Flexible aromatic disulfide monomers for high-perform-ance self-healable linear and cross-linked poly(urethane-urea) coatings. Polymer 2019, 166, 229–238. [Google Scholar] [CrossRef]
- Yu, K.; Xin, A.; Du, H.; Li, Y.; Wang, Q. Additive manufacturing of self-healing elastomers. NPG Asia Mater. 2019, 11, 7. [Google Scholar] [CrossRef] [Green Version]
- Ding, F.; Shi, X.; Wu, S.; Liu, X.; Deng, H.; Du, Y.; Li, H. Flexible Polysaccharide Hydrogel with pH-Regulated Reco-very of Self-Healing and Mechanical Properties. Macromol. Mater. Eng. 2017, 302, 1700221. [Google Scholar] [CrossRef]
- Li, C.; Wang, C.; Keplinger, C.; Zuo, J.; Jin, L.; Sun, Y.; Zheng, P.; Cao, Y. A highly stretchable autonomous self-healing elastomer. Nat. Chem. 2016, 8, 618–624. [Google Scholar] [CrossRef]
- Pettignano, A.; Häring, M.; Bernardi, L.; Tanchoux, N.; Quignard, F.; Díaz, D. Self-healing alginate–gelatin bioh-ydrogels based on dynamic covalent chemistry: Elucidation of key parameters. Mater. Chem. Front. 2017, 1, 73–79. [Google Scholar] [CrossRef] [Green Version]
- Yabuki, A.; Shiraiwa, T.; Fathona, I.W. pH-controlled self-healing polymer coatings with cellulose nanofibers providing an effective release of corrosion inhibitor. Corros. Sci. 2016, 103, 117–123. [Google Scholar] [CrossRef]
- Khan, A.; Huang, K.; Sarwar, M.G.; Rabnawaz, M. High modulus, fluorine-free self-healing anti-smudge coatings. Prog. Org. Coat. 2020, 145, 105703. [Google Scholar] [CrossRef]
- Gent, A.N.; Kawahara, S.; Zhao, J. Crystallization and Strength of Natural Rubber and Synthetic cis-1,4-Polyisoprene. Rubber Chem. Technol. 1998, 71, 668–678. [Google Scholar] [CrossRef]
- Li, W.; Liu, X.; Sun, A.; Wei, L.; Li, R.; He, H.; Yang, S.; Wang, S.; Niu, Y.; Li, Y. Exploring piperazine for intrinsic weather-proof, robust and self-healable poly(urethane urea) toward surface and tire protection. Polymer 2021, 227, 123829. [Google Scholar] [CrossRef]
- Liu, X.; Liu, X.; Li, W.; Ru, Y.; Li, Y.; Sun, A.; Wei, L. Engineered self-healable elastomer with giant strength and toughness via phase regulation and mechano-responsive self-reinforcing. Chem. Eng. J. 2021, 410, 128300. [Google Scholar] [CrossRef]
- Tang, M.; Zhang, R.; Li, S.; Zeng, J.; Luo, M.; Xu, Y.; Huang, G. Towards a Supertough Thermoplastic Polyisoprene Elastomer Based on a Biomimic Strategy. Angew. Chem. 2018, 130, 16062–16066. [Google Scholar] [CrossRef]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yi, P.; Chen, J.; Chang, J.; Wang, J.; Lei, Y.; Jing, R.; Liu, X.; Sun, A.; Wei, L.; Li, Y. Self-Healable, Strong, and Tough Polyurethane Elastomer Enabled by Carbamate-Containing Chain Extenders Derived from Ethyl Carbonate. Polymers 2022, 14, 1673. https://doi.org/10.3390/polym14091673
Yi P, Chen J, Chang J, Wang J, Lei Y, Jing R, Liu X, Sun A, Wei L, Li Y. Self-Healable, Strong, and Tough Polyurethane Elastomer Enabled by Carbamate-Containing Chain Extenders Derived from Ethyl Carbonate. Polymers. 2022; 14(9):1673. https://doi.org/10.3390/polym14091673
Chicago/Turabian StyleYi, Pengcheng, Jingrong Chen, Junyao Chang, Junbo Wang, Ying Lei, Ruobing Jing, Xingjiang Liu, Ailing Sun, Liuhe Wei, and Yuhan Li. 2022. "Self-Healable, Strong, and Tough Polyurethane Elastomer Enabled by Carbamate-Containing Chain Extenders Derived from Ethyl Carbonate" Polymers 14, no. 9: 1673. https://doi.org/10.3390/polym14091673