
Structure and Morphology of Crystalline Syndiotactic Polypropylene-Polyethylene Block Copolymers

 , , ,
, , ,  , , and
, , and 
Abstract
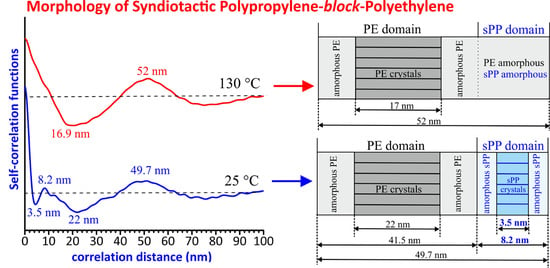
:
1. Introduction
2. Materials and Methods
3. Results and Discussion
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Hamley, I.W. Crystallization in Block Copolymers. Adv. Polym. Sci. 1999, 148, 113–137. [Google Scholar]
- Hamley, I.W. Introduction to Block Copolymers. In Development in Block Copolymer Science and Technology; Hamley, I.W., Ed.; John Wiley & Sons, Ltd.: Chichester, UK, 2004; p. 213. [Google Scholar]
- Loo, Y.L.; Register, R.A. Crystallization Within Block Copolymer Mesophases. In Development in Block Copolymer Science and Technology; Hamley, I.W., Ed.; John Wiley & Sons, Ltd.: Chichester, UK, 2004; pp. 213–244. [Google Scholar]
- Castillo, R.V.; Muller, A.J. Crystallization and morphology of biodegradable or biostable single and double crystalline block copolymers. Prog. Polym. Sci. 2009, 34, 516–560. [Google Scholar] [CrossRef]
- Muller, A.J.; Balsamo, V.; Arnal, M.L. Nucleation and Crystallization in Diblock and Triblock Copolymers. Adv. Polym. Sci. 2005, 190, 1–63. [Google Scholar]
- He, W.N.; Xu, J.T. Crystallization assisted self-assembly of semicrystalline block copolymers. Prog. Polym. Sci. 2012, 37, 1350–1400. [Google Scholar] [CrossRef]
- De Rosa, C.; Di Girolamo, R.; Malafronte, A.; Scoti, M.; Talarico, G.; Auriemma, R.; de Ballesteros, O. Polyolefins based crystalline block copolymers: Ordered nanostructures from control of crystallization. Polymer 2020, 196, 122423. [Google Scholar] [CrossRef]
- Van Horn, R.M.; Steffen, M.R.; O’Connor, D. Recent progress in block copolymer crystallization. Polym. Cryst. 2018, 1, e10039. [Google Scholar]
- Bates, F.S.; Fredrickson, G.H. Block Copolymer Thermodynamics: Theory and Experiment. Annu. Rev. Phys. Chem. 1990, 41, 525. [Google Scholar] [CrossRef]
- Lee, L.-B.W.; Register, R.A. Equilibrium Control of Crystal Thickness and Melting Point through Block Copolymerization. Macromolecules 2004, 37, 7278. [Google Scholar] [CrossRef]
- Loo, Y.L.; Register, R.A.; Ryan, A.J. Modes of Crystallization in Block Copolymer Microdomains: Breakout, Templated, and Confined. Macromolecules 2002, 35, 2365–2374. [Google Scholar] [CrossRef]
- Loo, Y.L.; Register, R.A.; Ryan, A.J.; Dee, G.T. Polymer crystallization confined in one, two, or three dimensions. Macromolecules 2001, 34, 8968. [Google Scholar] [CrossRef]
- Nojima, S.; Ono, M.; Ashida, T. Crystallization of block copolymers II. Morphological study of poly(ethylene glycol)-poly(ε-caprolactone) block copolymers. Polym. J. 1992, 24, 1271. [Google Scholar] [CrossRef]
- Sun, L.; Liu, Y.; Zhu, L.; Hsiao, B.S.; Avila-Orta, C.A. Self-assembly and crystallization behavior of a double-crystalline polyethylene-block-poly(ethylene oxide) diblock copolymer. Polymer 2004, 45, 8181. [Google Scholar] [CrossRef]
- Castillo, R.V.; Muller, A.J.; Lin, M.C.; Chen, H.L.; Jeng, U.S.; Hillmyer, M.A. Confined crystallization and morphology of melt segregated PLLA-b-PE and PLDA-b-PE diblock copolymers. Macromolecules 2008, 41, 6154. [Google Scholar] [CrossRef]
- Myers, S.B.; Register, R.A. Crystalline-Crystalline Diblock Copolymers of Linear Polyethylene and Hydrogenated Polynorbornene. Macromolecules 2008, 41, 6773. [Google Scholar] [CrossRef]
- De Rosa, C.; Di Girolamo, R.; Auriemma, F.; D’Avino, M.; Talarico, G.; Cioce, C.; Scoti, M.; Coates, G.W.; Lotz, B. Oriented Microstructures of Crystalline−Crystalline Block Copolymers Induced by Epitaxy and Competitive and Confined Crystallization. Macromolecules 2016, 49, 5576–5586. [Google Scholar] [CrossRef] [Green Version]
- De Rosa, C.; Malafronte, A.; Di Girolamo, R.; Auriemma, F.; Scoti, M.; Ruiz de Ballesteros, O.; Coates, G.W. Morphology of Isotactic Polypropylene−Polyethylene Block Copolymers Driven by Controlled Crystallization. Macromolecules 2020, 53, 10234–10244. [Google Scholar] [CrossRef]
- De Rosa, C.; Di Girolamo, R.; Cicolella, A.; Talarico, G.; Scoti, M. Double Crystallization and Phase Separation in Polyethylene—Polypropylene Di-Block Copolymers. Polymers 2021, 13, 2589. [Google Scholar] [CrossRef]
- Cano, L.; Builes, D.H.; Tercjak, A. Morphological and mechanical study of nanostructured epoxy systems modified with amphiphilic poly(ethylene oxide-b-propylene oxide-b-ethylene oxide)triblock copolymer. Polymer 2014, 55, 738–745. [Google Scholar] [CrossRef]
- Builes, D.H.; Hernández-Ortiz, J.P.; Corcuera, M.A.; Mondragon, I.; Tercjak, A. Effect of poly(ethylene oxide) homopolymer and two different poly(ethylene oxide-b-poly(propylene oxide)-b-poly(ethylene oxide) triblock copolymers on morphological, optical, and mechanical properties of nanostructured unsaturated polyester. ACS Appl. Mater. Interfaces 2014, 6, 1073–1081. [Google Scholar] [CrossRef]
- Tercjak, A.; Gutierrez, J.; Barud, H.S.; Domeneguetti, R.R.; Ribeiro, S.J.L. Nano- and macroscale structural and mechanical properties of in situ synthesized bacterial cellulose/PEO-b-PPO-b-PEO biocomposites. ACS Appl. Mater. Interfaces 2015, 7, 4142–4150. [Google Scholar] [CrossRef]
- Carrasco-Hernandez, S.; Gutierrez, J.; Cano, L.; Tercjak, A. Thermal and optical behavior of poly(ethylene-b-ethylene oxide) block copolymer dispersed liquid crystals blends. Eur. Polym. J. 2016, 74, 148–157. [Google Scholar] [CrossRef]
- Palacios, J.K.; Tercjak, A.; Liu, G.; Wang, D.; Zhao, J.; Hadjichristidis, N.; Müller, A.J. Trilayered Morphology of an ABC Triple Crystalline Triblock Terpolymer. Macromolecules 2017, 50, 7268–7281. [Google Scholar] [CrossRef]
- Carrasco-Hernandez, S.; Gutierrez, J.; Tercjak, A. PE-b-PEO block copolymer nanostructured thermosetting systems as template for TiO2 nanoparticles. Eur. Polym. J. 2017, 94, 87–98. [Google Scholar] [CrossRef]
- Matxinandiarena, E.; Múgica, A.; Tercjak, A.; Ladelta, V.; Zapsas, G.; Hadjichristidis, N.; Cavallo, D.; Flores, A.; Müller, A. Sequential Crystallization and Multicrystalline Morphology in PE- b-PEO- b-PCL- b-PLLA Tetrablock Quarterpolymers. Macromolecules 2021, 54, 7244–7257. [Google Scholar] [CrossRef]
- Cohen, R.E.; Cheng, P.L.; Douzinas, K.C.; Kofinas, P.; Berney, C.V. Path-dependent Morphologies of a Diblock Copolymer of Polystyrene/Hydrogenated Polybutadiene. Macromolecules 1990, 23, 324. [Google Scholar] [CrossRef]
- Douzinas, K.C.; Cohen, R.E. Chain Folding in EBEE Semicrystalline Diblock Copolymers. Macromolecules 1992, 25, 5030. [Google Scholar] [CrossRef]
- Cohen, R.E.; Bellare, A.; Drzewinski, M.A. Spatial Organization of Polymer Chains in a Crystallizable Diblock Copolymer of Polyethylene and Polystyrene. Macromolecules 1994, 27, 2321. [Google Scholar] [CrossRef]
- Kofinas, P.; Cohen, R.E. Morphology of Highly Textured Poly(ethylene)/Poly(ethylene-propylene) (E/EP) Semicrystalline Diblock Copolymers. Macromolecules 1994, 27, 3002. [Google Scholar] [CrossRef]
- Rangarajan, P.; Register, R.A.; Fetters, L.J. Morphology of Semicrystalline Block Copolymers of Ethylene-(Ethylene-alt-propylene). Macromolecules 1993, 26, 4640. [Google Scholar] [CrossRef]
- Rangarajan, P.; Register, R.A.; Adamson, D.H.; Fetters, L.J.; Bras, W.; Naylor, S.; Ryan, A.J. Dynamics of Structure Formation in Crystallizable Block Copolymers. Macromolecules 1995, 28, 1422. [Google Scholar] [CrossRef]
- Rangarajan, P.; Register, R.A.; Fetters, L.J.; Bras, W.; Naylor, S.; Ryan, A.J. Crystallization of a Weakly Segregated Polyolefin Diblock Copolymer. Macromolecules 1995, 28, 4932. [Google Scholar] [CrossRef]
- Ryan, A.J.; Hamley, I.W.; Bras, W.; Bates, F.S. Structure Development in Semicrystalline Diblock Copolymers Crystallizing from the Ordered Melt. Macromolecules 1995, 28, 3860. [Google Scholar] [CrossRef]
- Quiram, D.J.; Register, R.A.; Marchand, G.R. Crystallization of Asymmetric Diblock Copolymers from Microphase-Separated Melts. Macromolecules 1997, 30, 4551. [Google Scholar] [CrossRef]
- Quiram, D.J.; Register, R.A.; Marchand, G.R.; Ryan, A.J. Dynamics of Structure Formation and Crystallization in Asymmetric Diblock Copolymers. Macromolecules 1997, 30, 8338. [Google Scholar] [CrossRef]
- Quiram, D.J.; Register, R.A.; Marchand, G.R.; Adamson, D.H. Chain Orientation in Block Copolymers Exhibiting Cylindrically Confined Crystallization. Macromolecules 1998, 31, 4891. [Google Scholar] [CrossRef]
- De Rosa, C.; Park, C.; Thomas, E.L.; Lotz, B. Microdomain patterns via directional eutectic solidification and epitaxy. Nature 2000, 405, 433–437. [Google Scholar] [CrossRef]
- Park, C.; De Rosa, C.; Fetters, L.J.; Thomas, E.L. Influence of an Oriented Glassy Cylindrical Microdomain Structure on the Morphology of Crystallizing Lamellae in a Semicrystalline Block Terpolymer. Macromolecules 2000, 33, 7931. [Google Scholar] [CrossRef]
- De Rosa, C.; Park, C.; Lotz, B.; Wittmann, J.C.; Fetters, L.J.; Thomas, E.L. Control of Molecular and Microdomain Orientation in a Semicrystalline Block Copolymer Thin Film by Epitaxy. Macromolecules 2000, 33, 4871. [Google Scholar] [CrossRef]
- Park, C.; De Rosa, C.; Fetters, L.J.; Lotz, B.; Thomas, E.L. Alteration of Classical Microdomain Patterns of Block Copolymers by Degenerate Epitaxy. Adv. Mater. 2001, 13, 724. [Google Scholar] [CrossRef]
- Park, C.; De Rosa, C.; Lotz, B.; Fetters, L.J.; Thomas, E.L. Molecular and Microdomain Orientation in Semicrystalline Block Copolymer Thin Films by Directional Crystallization of the Solvent and Epitaxy. Macromol. Chem. Phys. 2003, 204, 1514. [Google Scholar] [CrossRef]
- Tian, J.; Hustad, P.D.; Coates, G.W. A New Catalyst for Highly Syndiospecific Living Olefin Polymerization: Homopolymers and Block Copolymers from Ethylene and Propylene. J. Am. Chem. Soc. 2001, 123, 5134. [Google Scholar] [CrossRef] [PubMed]
- Coates, G.W.; Hustad, P.D.; Reinartz, S. Catalysts for the Living Insertion Polymerization of Alkenes: Access to New Polyolefin Architectures Using Ziegler—Natta Chemistry. Angew. Chem. Int. Ed. 2002, 41, 2236. [Google Scholar] [CrossRef]
- Domski, G.J.; Rose, J.M.; Coates, G.W.; Bolig, A.D.; Brookhart, M. Living alkene polymerization: New methods for the precision synthesis of polyolefins. Prog. Polym. Sci. 2007, 32, 30. [Google Scholar] [CrossRef]
- Makio, H.; Terao, H.; Iwashita, A.; Fujita, T. FI Catalysts for Olefin Polymerization, A Comprehensive Treatment. Chem. Rev. 2011, 111, 2363. [Google Scholar] [CrossRef]
- De Rosa, C.; Di Girolamo, R.; Talarico, G. Expanding the Origin of Stereocontrol in Propene Polymerization Catalysis. ACS Catal. 2016, 6, 3767–3770. [Google Scholar] [CrossRef]
- De Rosa, C.; Auriemma, F.; Di Girolamo, R.; Aprea, R.; Thierry, A. Selective Gold Deposition on a Nanostructured Block Copolymer Film Crystallized by Epitaxy. Nano Res. 2011, 4, 241. [Google Scholar] [CrossRef]
- De Rosa, C.; Di Girolamo, R.; Auriemma, F.; Talarico, G.; Scarica, C.; Malafronte, A.; Scoti, M. Controlling Size and Orientation of Lamellar Microdomains in Crystalline Block Copolymers. ACS Appl. Mater. Interfaces 2017, 9, 31252–31259. [Google Scholar] [CrossRef]
- Ruokolainen, J.; Mezzenga, R.; Fredrickson, G.H.; Kramer, E.J.; Hustad, P.D.; Coates, G.W. Morphology and Thermodynamic Behavior of Syndiotactic Polypropylene-Poly(ethylene-co-propylene) Block Polymers Prepared by Living Olefin Polymerization. Macromolecules 2005, 38, 851. [Google Scholar] [CrossRef]
- Domski, G.J.; Eagan, J.M.; De Rosa, C.; Di Girolamo, R.; LaPointe, A.M.; Lobkovsky, E.B.; Talarico, G.; Coates, G.W. Combined Experimental and Theoretical Approach for Living and Isoselective Propylene Polymerization. ACS Catal. 2017, 7, 6930. [Google Scholar] [CrossRef] [Green Version]
- Eagan, J.M.; Xu, J.; Di Girolamo, R.; Thurber, C.M.; Macosko, C.W.; LaPointe, A.M.; Bates, F.S.; Coates, G.W. Combining polyethylene and polypropylene: Enhanced performance with PE/iPP multiblock polymers. Science 2017, 355, 814–816. [Google Scholar] [CrossRef] [Green Version]
- Xu, J.; Eagan, J.M.; Kim, S.-S.; Pan, S.; Lee, B.; Klimovica, K.; Jin, K.; Lin, T.-W.; Howard, M.J.; Ellison, C.J.; et al. Compatibilization of Isotactic Polypropylene (iPP) and High-Density Polyethylene (HDPE) with iPP-PE Multiblock Copolymers. Macromolecules 2018, 51, 8585–8596. [Google Scholar] [CrossRef] [Green Version]
- De Rosa, C.; Auriemma, F.; Malafronte, A.; Scoti, M. Crystal structures and polymorphism of polymers: Influence of defects and disorder. Polym. Cryst. 2018, 1, e10015. [Google Scholar] [CrossRef]
- De Rosa, C.; Scoti, M.; Di Girolamo, R.; Ruiz de Ballesteros, O.; Auriemma, F.; Malafronte, A. Polymorphism in polymers: A tool to tailor material’s properties. Polym. Cryst. 2020, 3, e10101. [Google Scholar] [CrossRef]
- De Rosa, C.; Auriemma, F. Structure and physical properties of syndiotactic polypropylene: A highly crystalline thermoplastic elastomer. Prog. Polym. Sci. 2006, 31, 145. [Google Scholar] [CrossRef]
- De Rosa, C.; Scoti, M.; Auriemma, F.; Ruiz de Ballesteros, O.; Talarico, G.; Malafronte, A.; Di Girolamo, R. Mechanical Properties and Morphology of Propene−Pentene Isotactic Copolymers. Macromolecules 2018, 51, 3030. [Google Scholar] [CrossRef]
- De Rosa, C.; Ruiz de Ballesteros, O.; Auriemma, F.; Talarico, G.; Scoti, M.; Di Girolamo, R.; Malafronte, A.; Piemontesi, F.; Liguori, D.; Camurati, I.; et al. Crystallization Behavior of Copolymers of Isotactic Poly(1-butene) with Ethylene from Ziegler−Natta Catalyst: Evidence of the Blocky Molecular Structure. Macromolecules 2019, 52, 9114. [Google Scholar] [CrossRef]
- Auriemma, F.; De Rosa, C.; Di Girolamo, R.; Malafronte, A.; Scoti, M.; Mitchell, G.R.; Esposito, S. Time-Resolving Study of Stress-Induced Transformations of Isotactic Polypropylene through Wide Angle X-ray Scattering Measurements. Polymers 2018, 10, 162. [Google Scholar] [CrossRef] [Green Version]
- Auriemma, F.; De Rosa, C.; Di Girolamo, R.; Malafronte, A.; Scoti, M.; Cipullo, R. Yield behavior of random copolymers of isotactic polypropylene. Polymer 2017, 129, 235. [Google Scholar] [CrossRef]
- De Rosa, C.; Scoti, M.; Ruiz de Ballesteros, O.; Di Girolamo, R.; Auriemma, F.; Malafronte, A. Propylene−Butene Copolymers: Tailoring Mechanical Properties from Isotactic Polypropylene to Polybutene. Macromolecules 2020, 53, 4407. [Google Scholar] [CrossRef]
- De Rosa, C.; Ruiz de Ballesteros, O.; Di Girolamo, R.; Malafronte, A.; Auriemma, F.; Talarico, G.; Scoti, M. The blocky structure of Ziegler–Natta “random” copolymers: Myths and experimental evidence. Polym. Chem. 2020, 11, 34. [Google Scholar] [CrossRef]
- De Rosa, C.; Scoti, M.; Auriemma, F.; Ruiz de Ballesteros, O.; Talarico, G.; Di Girolamo, R.; Cipullo, R. Relationships among lamellar morphology parameters, structure and thermal behavior of isotactic propene-pentene copolymers: The role of incorporation of comonomeric units in the crystals. Eur. Polym. J. 2018, 103, 251. [Google Scholar] [CrossRef]
- Scoti, M.; Di Girolamo, R.; Giusto, G.; De Stefano, F.; Auriemma, F.; Malafronte, A.; Talarico, G.; De Rosa, C. Mechanical Properties and Elastic Behavior of Copolymers of Syndiotactic Polypropylene with 1-Hexene and 1-Octene. Macromolecules 2021, 54, 6810. [Google Scholar] [CrossRef]
- Scoti, M.; Di Girolamo, R.; De Stefano, F.; Giordano, A.; Malafronte, A.; Talarico, G.; Cipullo, R.; De Rosa, C. Synthesis, structure and properties of copolymers of syndiotactic polypropylene with 1-hexene and 1-octene. Polym. Chem. 2021, 12, 5815. [Google Scholar] [CrossRef]
- Scoti, M.; De Stefano, F.; Di Girolamo, R.; Talarico, G.; Malafronte, A.; De Rosa, C. Crystallization of Propene-Pentene Isotactic Copolymers as Indicator of the General View of the Crystallization Behavior of Isotactic Polypropylene. Macromolecules 2022, 55, 241–251. [Google Scholar] [CrossRef]
- Brandrup, J.; Immergut, E.H.; Grulke, E.A. (Eds.) Polymer Handbook, 4th ed.; John Wiley & Sons: Hoboken, NJ, USA, 2003. [Google Scholar]
- Bergmann, A.; Orthaber, D.; Scherf, G.; Glatter, O. Improvement of SAXS measurements on Kratky slit systems by Göbel mirrors and imaging-plate detectors. J. Appl. Crystallogr. 2000, 33, 869–875. [Google Scholar] [CrossRef]
- Kratky, O.; Stabinger, H. X-ray small angle camera with block-collimation system an instrument of colloid research. Colloid Polym. Sci. 1984, 262, 345–360. [Google Scholar] [CrossRef]
- De Rosa, C.; Corradini, P. Crystal Structure of Syndiotactic Polypropylene. Macromolecules 1993, 26, 5711–5718. [Google Scholar] [CrossRef]
- Lotz, B.; Lovinger, A.J.; Cais, R.E. Crystal structure and morphology of syndiotactic polypropylene single crystals. Macromolecules 1988, 21, 2375. [Google Scholar] [CrossRef]
- Bunn, C.W. The crystal structure of long-chain normal paraffin hydrocarbons. The “shape”of the CH2 group. Trans. Faraday Soc. 1939, 35, 482–490. [Google Scholar] [CrossRef]
- De Rosa, C.; Auriemma, F.; Vinti, V. Disordered polymorphic modifications of form I of syndiotactic polypropylene. Macromolecules 1997, 30, 4137–4146. [Google Scholar] [CrossRef]
- Hamley, I.W.; Fairclough, J.P.A.; Terrill, N.J.; Ryan, A.J.; Lipic, P.M.; Bates, F.S.; Towns-Andrews, E. Crystallization in Oriented Semicrystalline Diblock Copolymers. Macromolecules 1996, 29, 8835. [Google Scholar] [CrossRef]
- Hamley, I.W.; Fairclough, J.P.A.; Ryan, A.J.; Bates, F.S.; Towns-Andrews, E. Crystallization of Nanoscale-Confined Diblock Copolymer Chains. Polymer 1996, 37, 4425. [Google Scholar] [CrossRef]
- Roe, R.-J. Methods of X-ray and Neutron Scattering in Polymer Science; Oxford University Press: New York, NY, USA, 2000. [Google Scholar]
- Ruland, W. Small-angle scattering of two-phase systems: Determination and significance of systematic deviations from Porod’s law. J. Appl. Cryst. 1971, 4, 70. [Google Scholar] [CrossRef]
- Vonk, C.G. A general computer program for the processing of small-angle X-ray scattering data. J. Appl. Cryst. 1975, 8, 340. [Google Scholar] [CrossRef] [Green Version]
- Wittmann, J.C.; Lotz, B. Polymer Decoration: The Orientation of Polymer Folds as Revealed by The Crystallization of Polymer Vapors. J. Polym. Sci. Polym. Phys. Ed. 1985, 23, 205. [Google Scholar] [CrossRef]
- Bassett, G.A. A New Technique for Decoration of Cleavage and Slip Steps on Ionic Crystal Surfaces. Philos. Mag. 1958, 3, 1042. [Google Scholar] [CrossRef]
- Ayache, J.; Beaunier, L.; Boumendil, J.; Ehret, G.; Laub, D. Sample Preparation Handbook for Transmission Electron Microscopy—Techniques; Springer: Berlin/Heidelberg, Germany, 2010; Chapter 7; p. 279. [Google Scholar]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
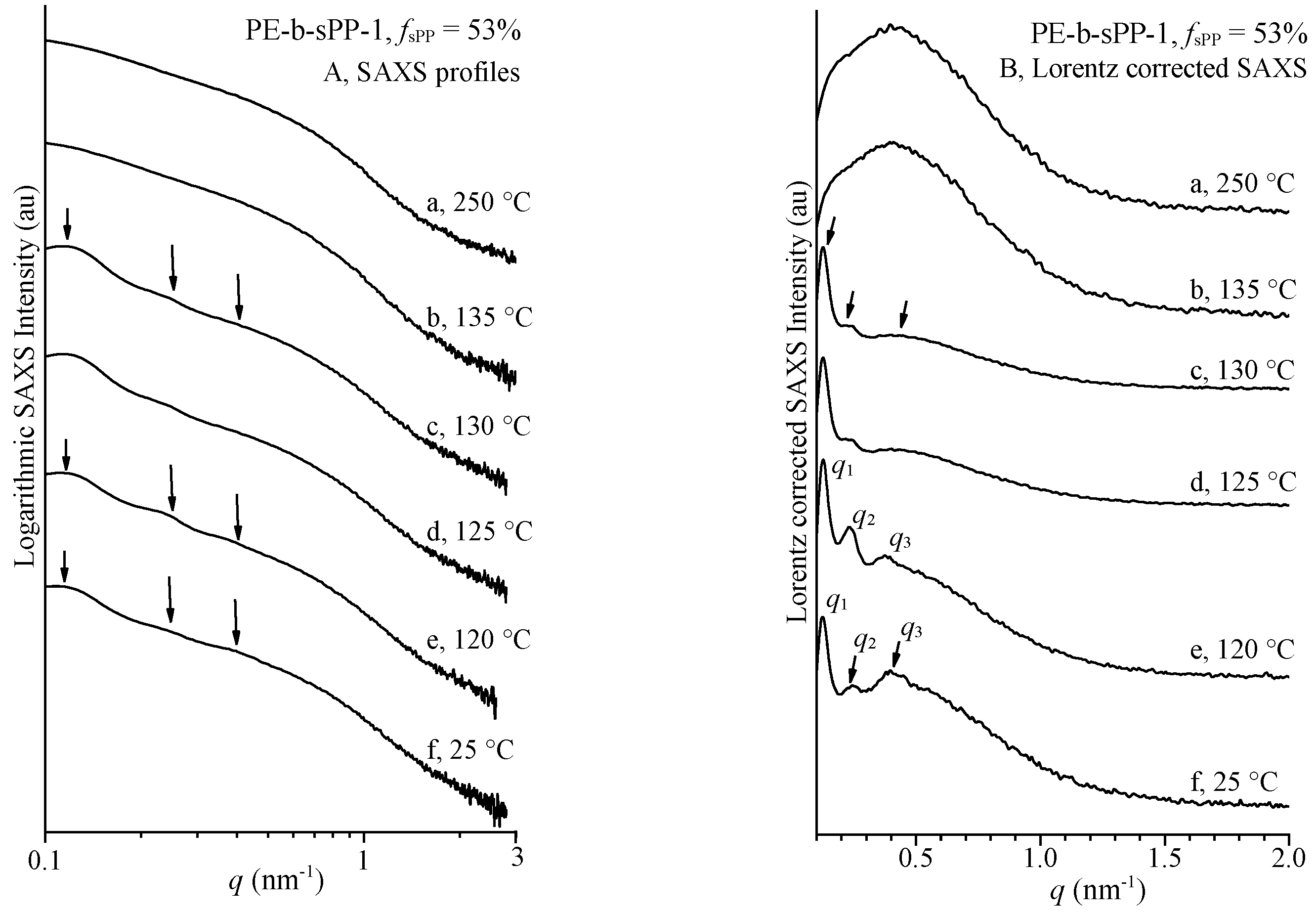{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | Mn (Da) | Mn(sPP) (Da) | Mn(PE) (Da) | wsPP (wt%) | fsPP (v/v%) | fPE (v/v%) | Mw /Mn |
|---|---|---|---|---|---|---|---|
| PE-b-sPP-1 | 20000 | 10200 | 9800 | 51 | 53 | 47 | 1.2 |
| PE-b-sPP-2 | 64000 | 46700 | 17300 | 73 | 75 | 25 | 1.2 |
| T (°C) | xc(WAXS) (%) | q1 (nm−1) | q2 (nm−1) | q3 (nm−1) | LB = 2π/q1 (nm) | lc * (nm) a | la* (nm) b | LCF (nm) | lc(CF) (nm) | 2la(CF) (nm) | LPE (nm) | LsPP (nm) |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 130 | 19 | 0.125 | 0.242 | 0.393 | 50.3 | 11.0 | 39.3 | 52.0 | 16.9 | 35.1 | ||
| 125 | 32 | 0.125 | 0.242 | 0.374 | 50.3 | 18.1 | 32.2 | 49.3 | 17.3 | 32.0 | ||
| 120 | 37 | 0.123 | 0.240 | 0.40 | 51.1 | 21.1 | 30.0 | 51.4 | 16.6 (PE) | 34.8 | ||
| 25 | 47 | 0.120 | 0.241 | 0.40 | 52.4 | 27.0 | 25.4 | 49.7 | 3.5 (sPP) 22 (PE) | 4.7 (sPP) 19.5 (PE) | 41.5 | 8.2 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Di Girolamo, R.; Cicolella, A.; Talarico, G.; Scoti, M.; De Stefano, F.; Giordano, A.; Malafronte, A.; De Rosa, C. Structure and Morphology of Crystalline Syndiotactic Polypropylene-Polyethylene Block Copolymers. Polymers 2022, 14, 1534. https://doi.org/10.3390/polym14081534
Di Girolamo R, Cicolella A, Talarico G, Scoti M, De Stefano F, Giordano A, Malafronte A, De Rosa C. Structure and Morphology of Crystalline Syndiotactic Polypropylene-Polyethylene Block Copolymers. Polymers. 2022; 14(8):1534. https://doi.org/10.3390/polym14081534
Chicago/Turabian StyleDi Girolamo, Rocco, Alessandra Cicolella, Giovanni Talarico, Miriam Scoti, Fabio De Stefano, Angelo Giordano, Anna Malafronte, and Claudio De Rosa. 2022. "Structure and Morphology of Crystalline Syndiotactic Polypropylene-Polyethylene Block Copolymers" Polymers 14, no. 8: 1534. https://doi.org/10.3390/polym14081534