
Ionic Liquids as Homogeneous Catalysts for Glycerol Oligomerization


Abstract
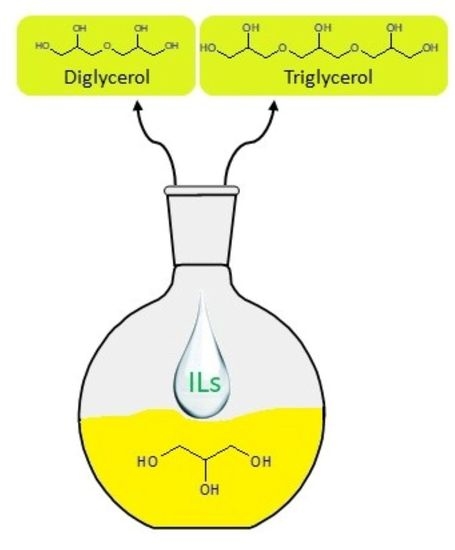
:
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Synthesis Ionic Liquids
2.3. Synthesis Oligoglycerols
2.4. Analysis
3. Results and Discussion
3.1. Characterization of ILs
3.2. Glycerin Oligomerization
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Saifuddin, N.M.; Hussein, R.; Ong, M. Sustainability of biodiesel production in Malaysia by production of bio-oil from crude glycerol using microwave pyrolysis: A review. Green Chem. Lett. Rev. 2018, 11, 135–157. [Google Scholar]
- OECD; Food and Agriculture Organization of the United Nations. OECD-FAO Agricultural Outlook 2017–2026; OECD: Paris, France, 2017. [Google Scholar] [CrossRef]
- Nanda, M.; Yuan, Z.; Qin, W.; Poirier, M.; Chunbao, X. Purification of crude glycerol using acidification: Effects of acid types and product characterization. Austin J. Chem. Eng. 2014, 1, 1–7. [Google Scholar]
- Anitha, M.; Kamarudin, S.; Kofli, N. The potential of glycerol as a value-added commodity. Chem. Eng. J. 2016, 295, 119–130. [Google Scholar] [CrossRef]
- Martin, A.; Richter, M. Oligomerization of glycerol—A critical review. Eur. J. Lipid Sci. Technol. 2011, 113, 100–117. [Google Scholar] [CrossRef]
- García-Sancho, C.; Moreno-Tost, R.; Mérida-Robles, J.M.; Santamaría-González, J.; Jiménez-López, A.; Torres, P.M. Etherification of glycerol to polyglycerols over MgAl mixed oxides. Catal. Today 2011, 167, 84–90. [Google Scholar] [CrossRef]
- Gholami, Z.; Lee, K.T.; Abdullah, A.Z. Glycerol etherification to polyglycerols using Ca1+xAlxLaxO3 composite catalysts in a solventless medium. J. Taiwan Inst. Chem. Eng. 2013, 44, 117–122. [Google Scholar] [CrossRef]
- Charles, G.; Clacens, J.-M.; Pouilloux, Y.; Barrault, J. Préparation de diglycérol et triglycérol par polymérisation directe du glycérol en présence de catalyseurs mésoporeux basiques. Oléagineux Corps Gras Lipides 2003, 10, 74–82. [Google Scholar] [CrossRef]
- Hensen, E.J.; Poduval, D.G.; Ligthart, D.M.; van Veen, J.R.; Rigutto, M.S. Quantification of strong Brønsted acid sites in aluminosilicates. J. Phys. Chem. C 2010, 114, 8363–8374. [Google Scholar] [CrossRef]
- Bender, J.; Jepkens, D.; Hüsken, H. Ionic liquids as phase-transfer catalysts: Etherification reaction of 1-Octanol with 1-Chlorobutane. Org. Process Res. Dev. 2010, 14, 716–721. [Google Scholar] [CrossRef]
- Ilavarasi Jeyamalar, J.; Krishnaveni, M.; Kannan, C. Synthesis and characterization of Ni-incorporated mesoporous silica material for its potential applications in oligomerization of glycerol. Phosphorus Sulfur Silicon Relat. Elem. 2022, 1–7. [Google Scholar] [CrossRef]
- Kansy, D.; Bosowska, K.; Czaja, K.; Poliwoda, A. The Formation of Glycerol Oligomers with Two New Types of End Groups in the Presence of a Homogeneous Alkaline Catalyst. Polymers 2019, 11, 144. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, H.J.; Seung, D.; Jung, K.S.; Kim, H.; Filimonov, I.N. Etherification of glycerol by isobutylene: Tuning the product composition. Appl. Catal. A Gen. 2010, 390, 235–244. [Google Scholar] [CrossRef]
- Zhao, C.; Chen, S.; Zhang, R.; Li, Z.; Liu, R.; Ren, B.; Zhang, S. Synthesis of propylene glycol ethers from propylene oxide catalyzed by environmentally friendly ionic liquids. Chin. J. Catal. 2017, 38, 879–888. [Google Scholar] [CrossRef]
- Zingg, A.; Winnefeld, F.; Holzer, L.; Pakusch, J.; Becker, S.; Gauckler, L. Adsorption of polyelectrolytes and its influence on the rheology, zeta potential, and microstructure of various cement and hydrate phases. J. Colloid Interface Sci. 2008, 323, 301–312. [Google Scholar] [CrossRef] [PubMed]
- Salehpour, S.; Dubé, M.A. Reaction Monitoring of Glycerol Step-Growth Polymerization Using ATR-FTIR Spectroscopy. Macromol. React. Eng. 2012, 6, 85–92. [Google Scholar] [CrossRef]
- Nguyen, R.; Galy, N.; Singh, A.K.; Paulus, F.; Stöbener, D.; Schlesener, C.; Sharma, S.K.; Haag, R.; Len, C. Simple and Efficient Process for Large Scale Glycerol Oligomerization by Microwave Irradiation. Catalysts 2017, 7, 123. [Google Scholar] [CrossRef]
- Nguyen, R.; Galy, N.; Alasmar, F.A.; Len, C. Ciągły przepływ wspomagany mikrofalami dla selektywnej oligomeryzacji glicerolu. Katalizatory 2021, 11, 166. [Google Scholar] [CrossRef]
- Medeiros, M.A.; Araujo, M.H.; Augusti, R.; De Oliveira, L.C.A.; Lago, R.M. Acid-Catalyzed Oligomerization of Glycerol Investigated by Electrospray Ionization Mass Spectrometry. J. Braz. Chem. Soc. 2009, 20, 1667–1673. [Google Scholar] [CrossRef]
- Bieńkowski, T.; Brodzik-Bieńkowska, A.; Danikiewicz, W. Complexes of bivalent metal cations in electrospray mass spectra of common organic compounds. J. Mass Spectrom. 2002, 37, 617–622. [Google Scholar] [CrossRef] [PubMed]
- D’Anna, F.; Vitale, P.; Noto, R. Determination of basic strength of aliphatic amines through ion pair formation in some ionic liquid solutions. J. Org. Chem. 2009, 74, 6224–6230. [Google Scholar] [CrossRef] [PubMed]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| IL Type | Amax | [I−] (%) | [HI] (%) | Hammett Function |
|---|---|---|---|---|
| H0 | ||||
| [C2-mim][Br] | 0.058 | 6.08 | 93.92 | 11.21 |
| [C2-mim][OAc] | 0.173 | 18.06 | 81.94 | 11.74 |
| [C12-mim][OAc] | 0.650 | 67.85 | 32.15 | 12.72 |
| [C14-mim][OAc] | 0.735 | 76.76 | 23.24 | 12.92 |
| [C12-tea][OAc] | 0.610 | 63.67 | 36.33 | 12.64 |
| [C12-mim][NaHPO4] | 0.495 | 51.69 | 48.31 | 12.43 |
| [C14-mim][NaHPO4] | 0.718 | 75.03 | 24.97 | 12.88 |
| Catalyst | Reaction Product Symbol | Reaction Rate Constant k [s−1] | Regression Coefficient R2 | Glycerols Conversion after 3 h [%] | Hammett Function H0 |
|---|---|---|---|---|---|
| Na2CO3 | PGL 1 | 1.56 × 10−4 | 0.991 | 86 | - |
| [C2-mim][Br] | PGL 8 | 1.74 × 10−4 | 0.749 | 89 | 11.21 |
| [C2-mim][OAc] | PGL 2 | 1.51 × 10−4 | 0.925 | 85 | 11.74 |
| [C12-mim][OAc] | PGL 6 | 1.44 × 10−4 | 0.952 | 83 | 12.72 |
| [C14-mim][OAc] | PGL 3 | 1.35 × 10−4 | 0.878 | 82 | 12.92 |
| [C12-tea][OAc] | PGL 5 | 2.10 × 10−4 | 0.936 | 92 | 12.64 |
| [C12-mim][NaHPO4] | PGL 7 | 1.49 × 10−4 | 0.971 | 85 | 12.43 |
| [C14-mim][NaHPO4] | PGL 4 | 1.14 × 10−4 | 0.994 | 76 | 12.88 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kansy, D.; Czaja, K.; Bosowska, K.; Groch, P. Ionic Liquids as Homogeneous Catalysts for Glycerol Oligomerization. Polymers 2022, 14, 1200. https://doi.org/10.3390/polym14061200
Kansy D, Czaja K, Bosowska K, Groch P. Ionic Liquids as Homogeneous Catalysts for Glycerol Oligomerization. Polymers. 2022; 14(6):1200. https://doi.org/10.3390/polym14061200
Chicago/Turabian StyleKansy, Dawid, Krystyna Czaja, Kornelia Bosowska, and Paweł Groch. 2022. "Ionic Liquids as Homogeneous Catalysts for Glycerol Oligomerization" Polymers 14, no. 6: 1200. https://doi.org/10.3390/polym14061200