
Thickness Dependence of Electronic Structure and Optical Properties of F8BT Thin Films

 ,
,  , and
, and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
3. Results and Discussion
3.1. UV-Vis Spectroscopy
3.2. Photoluminescence (PL) Spectroscopy
3.3. Density of State
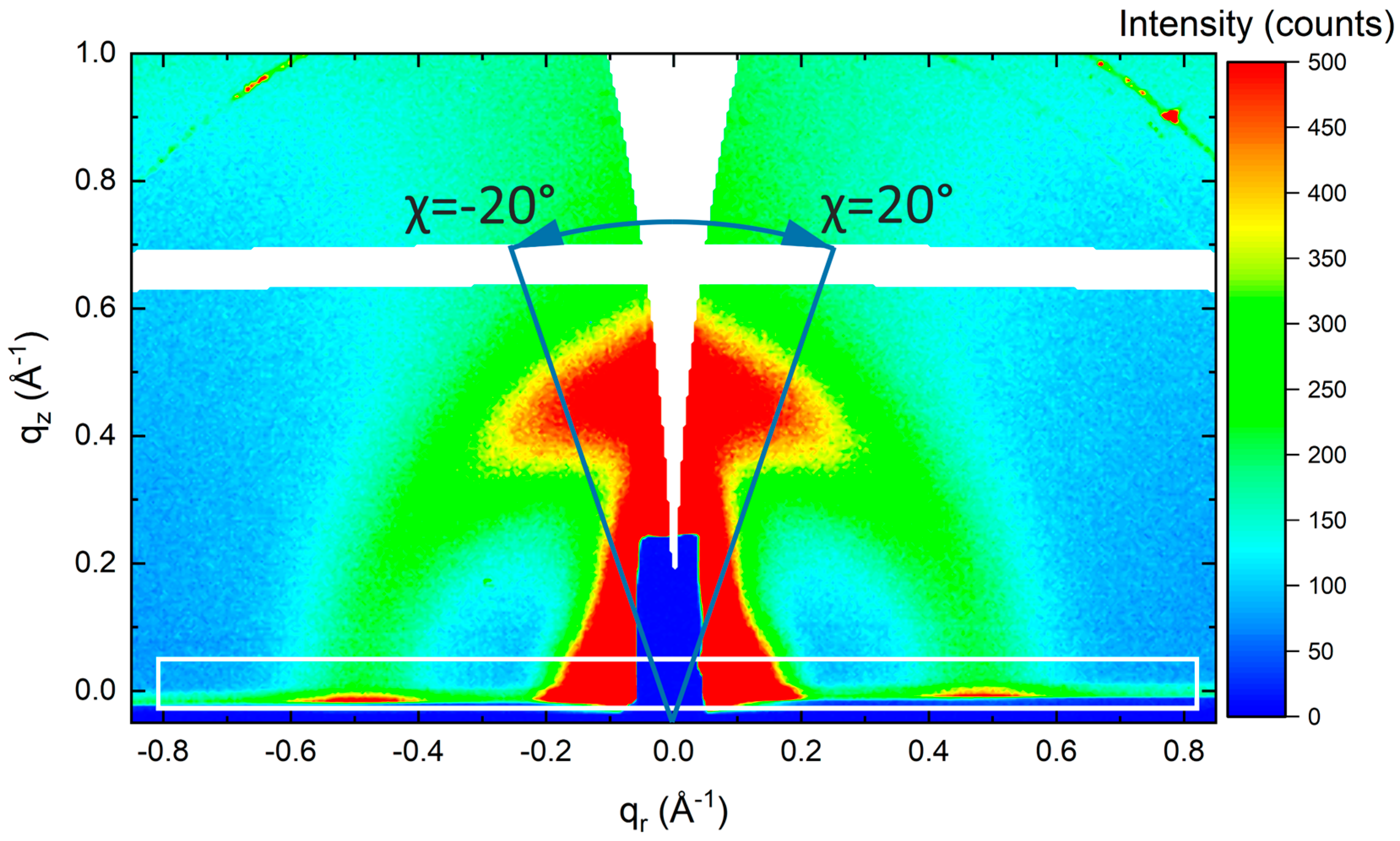
3.4. Structural Characterization—GIWAXS
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- NobelPrize.org. Available online: https://www.nobelprize.org/prizes/chemistry/ (accessed on 10 December 2021).
- Jaiswal, M.; Menon, R. Polymer electronic materials: A review of charge transport. Polym. Int. 2006, 55, 1371–1384. [Google Scholar] [CrossRef]
- Barbara, P.F.; Gesquiere, A.J.; Park, S.J.; Young, J.L. Single-molecule spectroscopy of conjugated polymers. Acc. Chem. Res. 2005, 38, 602–610. [Google Scholar] [CrossRef] [PubMed]
- Mei, J.; Bao, Z. Side chain engineering in solution-processable conjugated polymers. Chem. Mater. 2014, 26, 604–615. [Google Scholar] [CrossRef]
- An, C.; Zheng, Z.; Hou, J. Recent progress in wide bandgap conjugated polymer donors for high-performance nonfullerene organic photovoltaics. Chem. Commun. 2020, 56, 4750–4760. [Google Scholar] [CrossRef] [PubMed]
- Royakkers, J.; Bronstein, H. Macrocyclic Encapsulated Conjugated Polymers. Macromolecules 2021, 54, 1083–1094. [Google Scholar] [CrossRef]
- Liu, Y.; Wang, F.; Chen, J.; Wang, X.; Lu, H.; Qiu, L.; Zhang, G. Improved Transistor Performance of Isoindigo-Based Conjugated Polymers by Chemically Blending Strongly Electron-Deficient Units with Low Content to Optimize Crystal Structure. Macromolecules 2018, 51, 370–378. [Google Scholar] [CrossRef]
- Rasmussen, S.C. The path to conductive polyacetylene. Bull. Hist. Chem. 2014, 39, 64–72. [Google Scholar]
- Park, G.E.; Shin, J.; Lee, D.H.; Lee, T.W.; Shim, H.; Cho, M.J.; Pyo, S.; Choi, D.H. Acene-containing donor-acceptor conjugated polymers: Correlation between the structure of donor moiety, charge carrier mobility, and charge transport dynamics in electronic devices. Macromolecules 2014, 47, 3747–3754. [Google Scholar] [CrossRef]
- Neher, D. Polyfluorene homopolymers: Conjugated liquid-crystalline polymers for bright blue emission and polarized electroluminescence. Macromol. Rapid Commun. 2001, 22, 1365–1385. [Google Scholar] [CrossRef]
- Petrozza, A.; Fazzi, D.; Avilov, I.; Beljonne, D.; Friend, R.H.; Kim, J.S. Effects of polymer packing structure on photoinduced triplet generation and dynamics. J. Phys. Chem. C 2012, 116, 11298–11305. [Google Scholar] [CrossRef]
- Mamada, M.; Komatsu, R.; Adachi, C. F8BT Oligomers for Organic Solid-State Lasers. ACS Appl. Mater. Interfaces 2020, 12, 28383–28391. [Google Scholar] [CrossRef]
- Cheng, Z.; Javed, N.; Ocarroll, D.M. Optical and Electrical Properties of Organic Semiconductor Thin Films on Aperiodic Plasmonic Metasurfaces. ACS Appl. Mater. Interfaces 2020, 12, 35579–35587. [Google Scholar] [CrossRef] [PubMed]
- Bird, M.J.; Bakalis, J.; Asaoka, S.; Sirringhaus, H.; Miller, J.R. Fast Holes, Slow Electrons, and Medium Control of Polaron Size and Mobility in the da Polymer F8BT. J. Phys. Chem. C 2017, 121, 15597–15609. [Google Scholar] [CrossRef]
- Faria, G.C.; Faria, R.M. Molecular Structure and Dynamics of F8BT—Correlation with Opto-Electronic Properties. In Proceedings of the 2011 IEEE 14th International Symposium on Electrets ISE 14, Montpellier, France, 28–31 August 2011; pp. 101–102. [Google Scholar]
- Xiong, Y.; Peng, J.B.; Wu, H.B.; Wang, J. Improved performance of polymer light-emitting diodes with an electron transport emitter by post-annealing. Chinese Phys. Lett. 2009, 26, 097801. [Google Scholar]
- Bernardo, G.; Ferreira, Q.; Brotas, G.; Di Paolo, R.E.; Charas, A.; Morgado, J. Synergistic effect on the efficiency of polymer light-emitting diodes upon blending of two green-emitting polymers. J. Appl. Phys. 2010, 108, 014503. [Google Scholar] [CrossRef]
- Donley, C.L.; Zaumseil, J.; Andreasen, J.W.; Nielsen, M.M.; Sirringhaus, H.; Friend, R.H.; Kim, J.S. Effects of packing structure on the optoelectronic and charge transport properties in poly(9,9-di-n-octylfluorene-alt-benzothiadiazole). J. Am. Chem. Soc. 2005, 127, 12890–12899. [Google Scholar] [CrossRef]
- Clark, J.; Chang, J.F.; Spano, F.C.; Friend, R.H.; Silva, C. Determining exciton bandwidth and film microstructure in polythiophene films using linear absorption spectroscopy. Appl. Phys. Lett. 2009, 94, 2007–2010. [Google Scholar] [CrossRef]
- Spano, F.C. The spectral signatures of frenkel polarons in H- And J-aggregates. Acc. Chem. Res. 2010, 43, 429–439. [Google Scholar] [CrossRef]
- Urbánek, P.; Kuřitka, I.; Ševčík, J.; Toušková, J.; Toušek, J.; Nádaždy, V.; Nádaždy, P.; Végsö, K.; Šiffalovič, P.; Rutsch, R.; et al. An experimental and theoretical study of the structural ordering of the PTB7 polymer at a mesoscopic scale. Polym. Guildf. 2019, 169, 243–254. [Google Scholar] [CrossRef]
- Urbánek, P.; Kuřitka, I.; Daniš, S.; Toušková, J.; Toušek, J. Thickness threshold of structural ordering in thin MEH-PPV films. Polym. Guildf. 2014, 55, 4050–4056. [Google Scholar] [CrossRef]
- Urbánek, P.; Kuřitka, I. Thickness dependent structural ordering, degradation and metastability in polysilane thin films: A photoluminescence study on representative σ-conjugated polymers. J. Lumin. 2015, 168, 261–268. [Google Scholar] [CrossRef] [Green Version]
- Nádazdy, V.; Schauer, F.; Gmucová, K. Energy resolved electrochemical impedance spectroscopy for electronic structure mapping in organic semiconductors. Appl. Phys. Lett. 2014, 105, 142109. [Google Scholar] [CrossRef] [Green Version]
- Schauer, F.; Nádazdy, V.; Gmucová, K. Electrochemical impedance spectroscopy for study of electronic structure in disordered organic semiconductors—Possibilities and limitations. J. Appl. Phys. 2018, 123, 161590. [Google Scholar] [CrossRef]
- Bässler, H.; Kroh, D.; Schauer, F.; Nádaždy, V.; Köhler, A. Mapping the Density of States Distribution of Organic Semiconductors by Employing Energy Resolved–Electrochemical Impedance Spectroscopy. Adv. Funct. Mater. 2021, 31, 2007738. [Google Scholar] [CrossRef]
- Karki, A.; Wetzelaer, G.-J.A.H.; Reddy, G.N.M.; Nádaždy, V.; Seifrid, M.; Schauer, F.; Bazan, G.C.; Chmelka, B.F.; Blom, P.W.M.; Nguyen, T.-Q. Unifying Energetic Disorder from Charge Transport and Band Bending in Organic Semiconductors. Adv. Funct. Mater. 2019, 29, 1901109. [Google Scholar] [CrossRef]
- Athanasopoulos, S.; Schauer, F.; Nádaždy, V.; Weiß, M.; Kahle, F.-J.; Scherf, U.; Bässler, H.; Köhler, A. What is the Binding Energy of a Charge Transfer State in an Organic Solar Cell? Adv. Energy Mater. 2019, 9, 1900814. [Google Scholar] [CrossRef]
- Denis, J.C.; Ruseckas, A.; Hedley, G.J.; Matheson, A.B.; Paterson, M.J.; Turnbull, G.A.; Samuel, I.D.W.; Galbraith, I. Self-trapping and excited state absorption in fluorene homo-polymer and copolymers with benzothiadiazole and tri-phenylamine. Phys. Chem. Chem. Phys. 2016, 18, 21937–21948. [Google Scholar] [CrossRef] [Green Version]
- Spano, F.C.; Clark, J.; Silva, C.; Friend, R.H. Determining exciton coherence from the photoluminescence spectral line shape in poly(3-hexylthiophene) thin films. J. Chem. Phys. 2009, 130, 074904. [Google Scholar] [CrossRef]
- Spano, F.C.; Yamagata, H. Vibronic coupling in J-aggregates and beyond: A direct means of determining the exciton coherence length from the photoluminescence spectrum. J. Phys. Chem. B 2011, 115, 5133–5143. [Google Scholar] [CrossRef]
- Zahn, D.R.T.; Gavrila, G.N.; Gorgoi, M. The transport gap of organic semiconductors studied using the combination of direct and inverse photoemission. Chem. Phys. 2006, 325, 99–112. [Google Scholar] [CrossRef]
- Venkateshvaran, D.; Nikolka, M.; Sadhanala, A.; Lemaur, V.; Zelazny, M.; Kepa, M.; Hurhangee, M.; Kronemeijer, A.J.; Pecunia, V.; Nasrallah, I.; et al. Approaching disorder-free transport in high-mobility conjugated polymers. Nature 2014, 515, 384–388. [Google Scholar] [CrossRef] [Green Version]
- Botiz, I.; Stingelin, N. Influence of molecular conformations and microstructure on the optoelectronic properties of conjugated polymers. Materials 2014, 7, 2273–2300. [Google Scholar] [CrossRef] [Green Version]
- Noriega, R.; Rivnay, J.; Vandewal, K.; Koch, F.P.V.; Stingelin, N.; Smith, P.; Toney, M.F.; Salleo, A. A general relationship between disorder, aggregation and charge transport in conjugated polymers. Nat. Mater. 2013, 12, 1038–1044. [Google Scholar] [CrossRef]
- Sevcik, J.; Urbanek, P.; Skoda, D.; Jamatia, T.; Nadazdy, V.; Urbanek, M.; Antos, J.; Munster, L.; Kuritka, I. Energy resolved-electrochemical impedance spectroscopy investigation of the role of Al-doped ZnO nanoparticles in electronic structure modification of polymer nanocomposite LEDs. Mater. Des. 2021, 205, 109738. [Google Scholar] [CrossRef]
- Skoda, D.; Urbanek, P.; Sevcik, J.; Munster, L.; Antos, J.; Kuritka, I. Microwave-assisted synthesis of colloidal ZnO nanocrystals and their utilization in improving polymer light emitting diodes efficiency. Mater. Sci. Eng. B Solid-State Mater. Adv. Technol. 2018, 232–235, 22–32. [Google Scholar] [CrossRef]
- Skoda, D.; Urbanek, P.; Sevcik, J.; Munster, L.; Nadazdy, V.; Cullen, D.A.; Bazant, P.; Antos, J.; Kuritka, I. Colloidal cobalt-doped ZnO nanoparticles by microwave-assisted synthesis and their utilization in thin composite layers with MEH-PPV as an electroluminescent material for polymer light emitting diodes. Org. Electron. 2018, 59, 337–348. [Google Scholar] [CrossRef]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ghasemi, B.; Ševčík, J.; Nádaždy, V.; Végsö, K.; Šiffalovič, P.; Urbánek, P.; Kuřitka, I. Thickness Dependence of Electronic Structure and Optical Properties of F8BT Thin Films. Polymers 2022, 14, 641. https://doi.org/10.3390/polym14030641
Ghasemi B, Ševčík J, Nádaždy V, Végsö K, Šiffalovič P, Urbánek P, Kuřitka I. Thickness Dependence of Electronic Structure and Optical Properties of F8BT Thin Films. Polymers. 2022; 14(3):641. https://doi.org/10.3390/polym14030641
Chicago/Turabian StyleGhasemi, Bita, Jakub Ševčík, Vojtěch Nádaždy, Karol Végsö, Peter Šiffalovič, Pavel Urbánek, and Ivo Kuřitka. 2022. "Thickness Dependence of Electronic Structure and Optical Properties of F8BT Thin Films" Polymers 14, no. 3: 641. https://doi.org/10.3390/polym14030641