
Self-Attractive Semiflexible Polymers under an External Force Field

Istituto Applicazioni Calcolo, Consiglio Nazionale delle Ricerche (CNR), Via Amendola 122/D, 70126 Bari, Italy
Polymers 2022, 14(21), 4762; https://doi.org/10.3390/polym14214762
Submission received: 18 October 2022
/
Revised: 31 October 2022
/
Accepted: 2 November 2022
/
Published: 7 November 2022
(This article belongs to the Special Issue Feature Papers in Polymer Physics and Theory)
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:The dynamical response of a tethered semiflexible polymer with self-attractive interactions and subjected to an external force field is numerically investigated by varying stiffness and self-interaction strength. The chain is confined in two spatial dimensions and placed in contact with a heat bath described by the Brownian multi-particle collision method. For strong self-attraction the equilibrium conformations range from compact structures to double-stranded chains, and to rods when increasing the stiffness. Under the external field at small rigidities, the initial close-packed chain is continuously unwound by the force before being completely elongated. For double-stranded conformations the transition from the folded state to the open one is sharp being steeper for larger stiffnesses. The discontinuity in the transition appears in the force-extension relation, as well as in the probability distribution function of the gyration radius. The relative deformation with respect to the equilibrium case along the direction normal to the force is found to decay as the inverse of the applied force.
1. Introduction
The study of single polymers, such as, for example, DNA, filamentous actin, and microtubules under various flow conditions, has helped in understanding their dynamical and conformational properties [1]. The first investigations of the flow behavior of single DNA filaments [2] opened the way to a large variety of flow experiments which provided insight into the mechanisms regulating the dynamics. Several computational models have been studied which reveal to be very useful in understanding such systems. Single polymer studies give the chance of directly observing the microscopic conformations of individual chains close to equilibrium or under flow conditions, thus accessing non-equilibrium conformations.
In the case of biological filaments, their stiffness is closely related to their functions. For example, the rigidity of actin filaments is responsible for the mechanical properties of the cytoskeleton, and DNA is able to pack in the genome or inside a virus capsid thanks to its persistence length. Several works have investigated the equilibrium properties of semiflexible polymers [3,4,5,6,7,8,9,10]. The development of spectroscopic techniques and fluorescence microscopy provided insight into their non-equilibrium properties (for reviews see, e.g., Refs. [11,12]). Theoretical [13,14,15,16,17] and computational [18,19,20,21,22,23,24,25,26,27,28,29] studies helped in revealing and understanding novel dynamical, conformational, and rheological properties.
Among others, the worm-like chain model [30] proved to be accurate to describe the mechanical response of semiflexible polymers under specific conditions. Indeed, the main limitations of this model come from neglecting excluded volume effects and self-interactions between different polymer parts. The former are relevant, especially in two dimensions leading, for example, to the segregation of polymers [31]. The latter interactions, that are not relevant for strong applied fields or far from the folding temperature, are crucial for semiflexible chains with monomer–monomer interactions, such as poly(ethylene oxide) (PEO), DNA [32], or RNA [33] in poor-solvent condition [34]. Short-range attractive interactions lead to a large variety of conformations due to the competition of polymer stretching and collapse [35]. Previous experimental [36,37], theoretical, and numerical [38,39,40,41,42,43,44,45,46] studies have found that the mechanical response of self-interacting semiflexible polymers to an external stretching is very complicated. These investigations considered either chains with one of its ends grafted and the other one pulled by a force, or chains with both ends pulled away in opposite directions.
A large majority of studies has been performed in three dimensions but addressing the comprehension of stretched self-interacting polymers in two dimensions is also interesting for two main reasons: excluded-volume effects are relevant and hydrodynamic interactions can be neglected in the case of polymers strongly adsorbed on surfaces since the overall dynamics is dominated by the polymer–substrate interaction [47]. Two-dimensional realizations of these systems can, for example, be provided by DNA strongly adsorbed on a surface with one grafted end. Under these conditions the stretching of biopolymers is observed in systems with separation of biomolecules via nanochannels [48,49]. The effects of a uniform force field on two-dimensional semiflexible polymers have been considered both in experimental [47] and numerical [50,51] studies but neglecting self-attractions among monomers.
So far, a systematic study of polymers under poor-solvent condition in an external field is lacking. In the present work, the dynamical and conformational properties of a semiflexible filament, tethered by one of its ends and subjected to an external force field in two spatial dimensions, are numerically investigated. The polymer is modeled as a self-avoiding worm-like chain with self-attraction among beads. Hydrodynamics is neglected since it is assumed that local polymer friction is uniquely fixed by its interaction with the adsorbing surface. For this reason the polymer is taken to be in contact with a Brownian heat bath. This is implemented by adopting the Brownian version [52] of the multi-particle collision dynamics [53,54]. By varying stiffness and self-interaction strength, different equilibrium conformations are found. For strong mutual attraction and relative low stiffness, the structure is compact. Increasing the chain rigidity promotes the formations of folded strands. The mechanical response of the polymer to the applied force depends on the equilibrium structure. At small rigidities the initial close-packed chain is continuously unwound by the external force field. The polymer shows bistable conformations before being completely elongated. When double-stranded chains form, a “first-order”-like phase transition to the open conformation is observed in the force-extension curve. Polymer configurations are characterized by considering the gyration tensor: it is found that the relative deformation with respect to the equilibrium case along the direction normal to the force, decays as the inverse of the applied force.
2. Model and Method
A linear chain of length L, made of beads of mass M, is considered in two spatial dimensions. Internal forces acting on beads are due to a potential which accounts for different contributions. Connected beads interact via the harmonic potential
where denotes the position vector of the ith bead (), is the average bond length, and the elasticity is controlled by . The parameter is chosen in order to preserve on the average the total contour length . Chain stiffness of the polymer is introduced via the bending potential
where is the bending rigidity and is the angle between two consecutive bond vectors. Non-bonded pair interactions are modeled by the Lennard–Jones potential
where is the distance between two non-consecutive beads. A strongly attractive regime corresponds to energies , which determine compact structures. In the opposite limit of weak self-attraction, swollen chain configurations can be observed. Here is the thermal energy, T is the temperature, and is Boltzmann’s constant. The parameters and are varied in the present study, keeping fixed the temperature, to obtain different equilibrium conformations as later shown. In the following, for the sake of clarity, chain stiffness is characterized in terms of the length . In the worm-like chain limit, when the Lennard–Jones potential is negligible, this length corresponds to the polymer persistence length [55]. However, in the present model this is not strictly true due to the coexistence of different length and energy scales [56]. Finally, in order to consider external stretching of the chain, a constant force F acts on every bead of the polymer. This force is directed along the x-direction of the Cartesian reference frame and corresponds to an external potential given by
The external field could be a gravitational or uniform flow field. Newton’s equations of motion of beads are integrated by the velocity-Verlet algorithm with time step [57,58].
The chain is coupled to a Brownian heat bath which is implemented by using the Brownian multi-particle collision (B-MPC) method [52,54,59] without taking into account hydrodynamic interactions. Here, we adopt the computationally efficient version proposed in Ref. [52]. In this algorithm, every bead undergoes stochastic collisions with a virtual particle of mass M to simulate the interaction with a fluid volume surrounding the bead. The momenta of such phantom particles are Maxwell–Boltzmann distributed with variance and zero mean. The collision process is implemented via the stochastic rotation dynamics of the MPC method [54,60,61]. This corresponds to randomly rotate the relative velocity of a polymer bead, with respect to the center-of-mass velocity of the bead and its related phantom particle, by angles . Collisions occur at time intervals being .
Simulations are performed with the choices , , with time unit , , , , , and . The value of ensures that the polymer length L is constant within for all systems.
3. Numerical Results
Polymers are initialized with beads randomly aligned along the x-direction and allowed to equilibrate. The position of the first bead is fixed at the origin of the Cartesian reference frame while no orientation is enforced for the first bond. When taking into account the action of the uniform force field, simulations are started from the equilibrium configurations of chains and run until reaching steady states during which average quantities are computed. We consider semiflexible polymers with values of the bending rigidity , such that , and interaction energies .
3.1. Equilibrium Polymer Conformations
In this Section, the equilibrium properties of polymers are obtained and characterized by varying the bending rigidity and the interaction energy. When considering the value , non-bonded interactions are negligibly small and the model corresponds to the worm-like chain model [30], as shown in the following. In this case, the filament assumes a swollen configuration with spatial correlations, in the direction of the chain tangent, on a length scale given by the persistence length. A different scenario occurs when non-bonded interactions become relevant. Equilibrium configurations for and different values of the length are shown in Figure 1.
For the smallest value (Figure 1a) the chain has a globule structure which is very compact. Increasing the stiffness promotes the formation of folded bundles. A configuration with five rod-like strands is shown in Figure 1b for . The energy penalty, which is proportional to and increases with the bending angle at turning points, is compensated by the energy gain from bead–bead attractions. The number of strands diminishes when increasing . A structure formed by two facing strands is observed at (Figure 1c). For this value of , the average bending energy diminishes since the number of turning points reduces, and the average Lennard–Jones energy increases. A further increase in chain stiffness induces the formation of hairpin conformations (Figure 1d for ). This causes a second rise in the bending energy whose energetic penalty can still be compensated by the mutual attraction between monomers. Finally, at the polymer cannot sustain any closed configuration and a rod-like structure is observed for values . In this latter range, the average value of exhibits a sharp increase while the average bending energy decreases.
In order to characterize the conformations of chains, it is useful to consider the root-mean-square values of the end-to-end distance , where , and of the gyration radius . By computing the gyration tensor
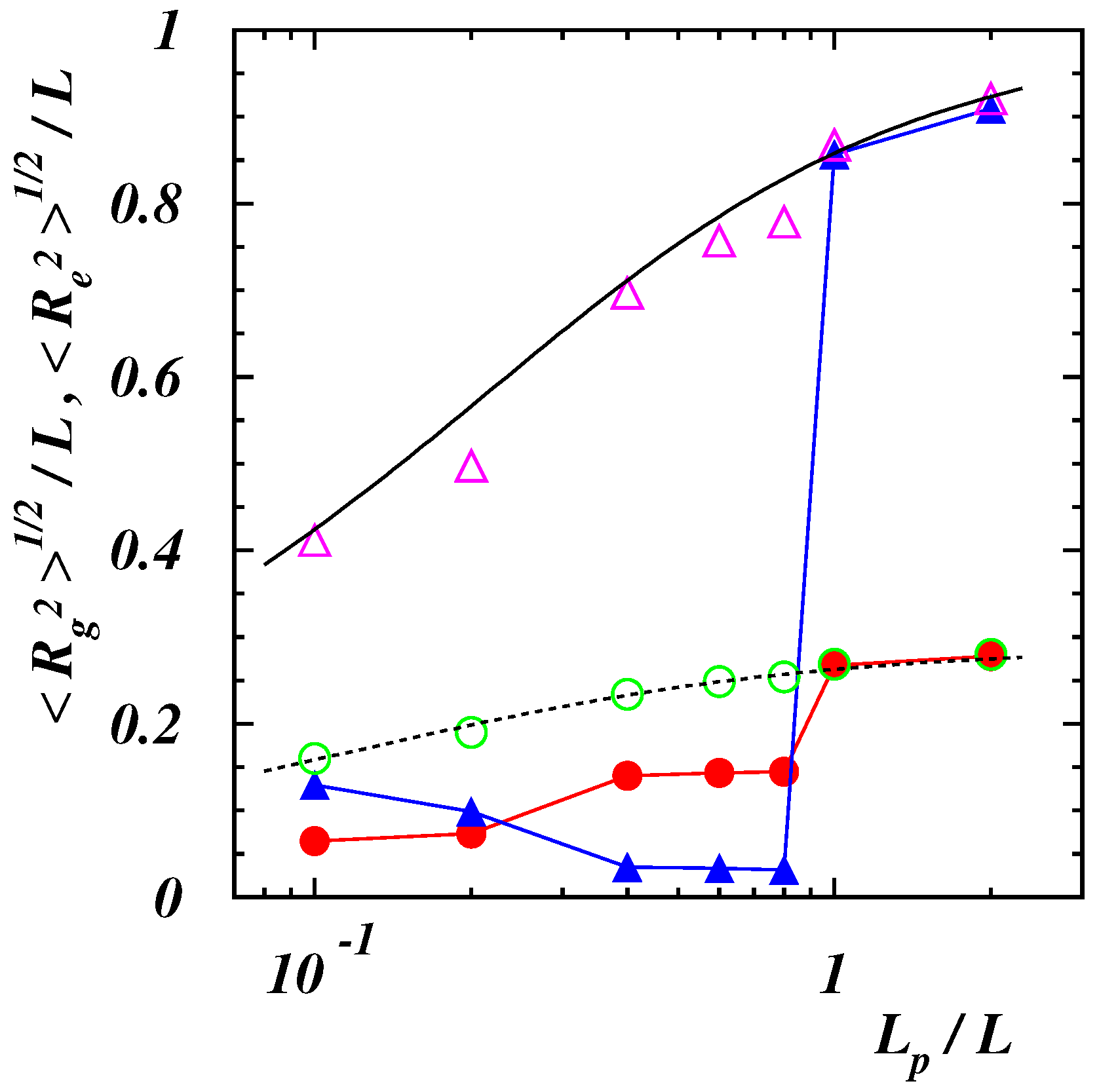
where is the position of the i-th monomer in the center-of-mass reference frame of the chain and the Greek index denotes the Cartesian component, the gyration radius can be obtained as . The computed values of and for the two values of as functions of the dimensionless length are presented in Figure 2.
For the smallest value the numerical results show a quantitative agreement with the theoretical predictions for a continuous semiflexible chain [55]
This confirms that the self-interaction energy is negligible for this choice of and the polymer behaves as a worm-like chain. The behavior is different for the highest value of the energy . The end-to-end distance is smaller than in the previous case and decreases to reach its minimum value when the chain consists of two strands folded on each other (). Then, jumps to values comparable to those of semiflexible polymers at . The average gyration radius is at a minimum when compact conformations are observed (), then increases to a value which remains constant as long as the chain consists of two strands, and finally reaches the equilibrium values of worm-like chains when the polymer assumes a rod-like structure.
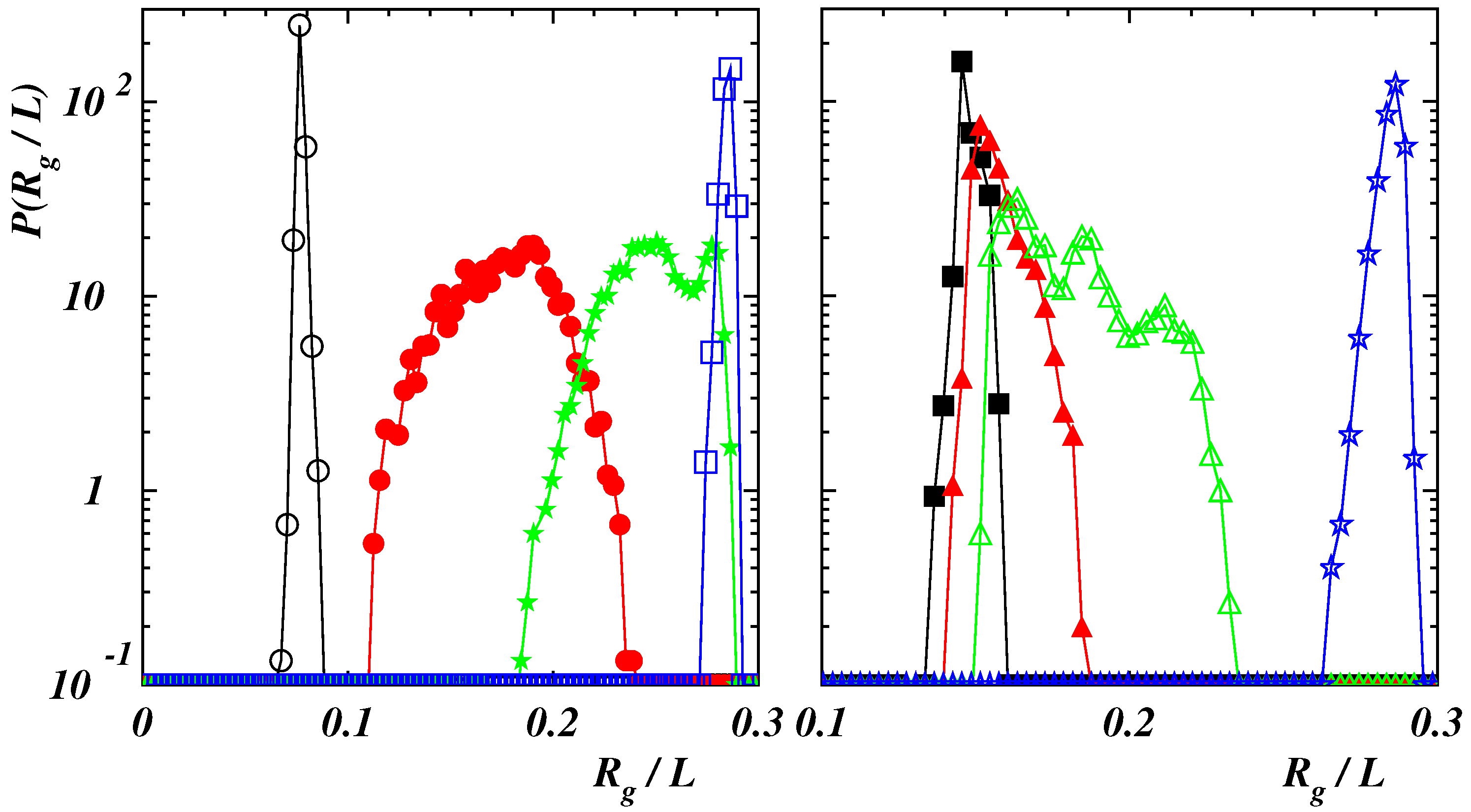
Normalized probability distribution functions (PDFs) of the polymer gyration radius are depicted in Figure 3 under equilibrium conditions.
For the highest value of the interaction energy , when the polymer is compact, curves are very narrow corresponding to the fact that the chain global conformation does not change significantly in time. The two curves with almost overlap with peaks located at . When considering double-stranded chains, the curve at is broader since the chain fluctuates along its length. The peak is at as in the case with where the PDF is narrower since the structure is quite rigid. Finally, when the polymer assumes a rod-like conformation (), the position of the PDF peak moves to . For a comparison two PDFs in the case of weak self-attraction () are also presented in the figure. Curves are broader than in the previous case due to the fact that chains are more prone to fluctuate since the mutual attraction is negligible. The peaks are located at larger values of with respect to the case with , for the same stiffness, corresponding to more elongated structures.
3.2. Polymer Stretching in Uniform Force Field
When the polymer is subject to the external force, it is stretched along the direction of the force. In order to characterize the elongation of the chain, the average deficit length-ratio as a function of the applied force is considered. is the average extension of the chain along the force direction computed as the average value of the x-component of the end-to-end vector . When self-attraction is negligible, in the limit it results [47,62]
with . For quite strong force fields or very small bending rigidities the behavior does not depend on the stiffness and is given by [38,50,63]
where , as for flexible chains [64].
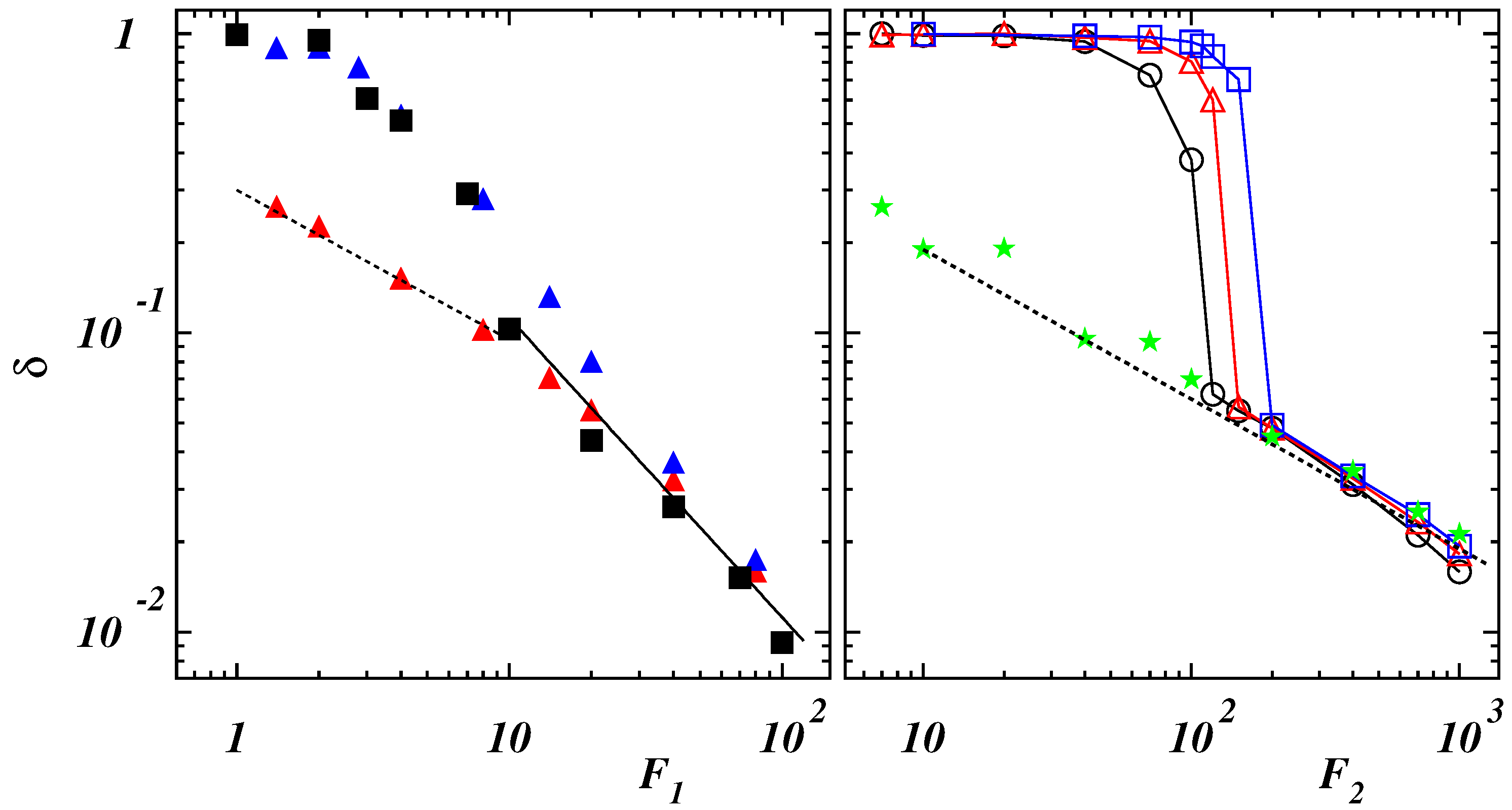
Different behaviors can be expected for self-interacting semiflexible polymers. When the filament is pulled at one end by a constant force, a sharp transition appears in the force vs. elongation curves [38] whose sharpness is enhanced by bending rigidity [42]. Simulations results of the present model are illustrated in Figure 4 as functions of applied force for different values of the ratio .
In case of , corresponding to compact initial states (see Figure 1), data collapse is obtained when plotting values of as functions of the dimensionless force (left panel of Figure 4). The initial structure is tilted in the direction of the force and only slightly deformed as long as . This can also be appreciated when considering the normalized PDFs of the gyration radius: in the case with and , the PDF exhibits a narrow peak (see Figure 5 (left panel)).
By increasing the force, the extension increases smoothly since the globule is partially unwound, similarly to what holds for single-stranded DNA and RNA [38,39]. A chain-and-blob [65] configuration can be observed where the blob at the end fluctuates in shape and size due to thermal fluctuations (see the Supplementary Video S1 for and ). The corresponding PDF broadens while still displaying a single peak which moves toward larger values of . At , the chain is stretched although, from time to time, the final part can be still folded due to self-attraction (see the Supplementary Video S2 for and ). The PDF of exhibits two peaks corresponding to fully elongated and partially bent conformational states which are stable for relatively long times to be clearly observed. This multi-peak feature is similar to that observed for pulled semiflexible polymers under poor-solvent condition [42,66,67] and proteins subject to a uniform flow [68]. By further increasing the force, the polymer is completely elongated with a narrow PDF of whose position shifts continuously to larger values of . The relation (9), observed once , indicates that the chain behaves as a semiflexible polymer under strong force. As a matter of comparison, we report also the results in the case when self-attraction is negligible for a similar bending rigidity (see the data for and in the left panel of Figure 4). The behavior at small force values is different with the deficit length-ratio decaying as , which is typical of semiflexible polymers without self-interaction. By increasing the force, the dependence (9) is recovered with the values of collapsing onto the ones for .
When the stiffness of the chain is such that a polymer exhibits a double-stranded conformation, the mechanical response to the external force is different as it can be seen in the right panel of Figure 4 where the average deficit length-ratio is plotted as a function of the dimensionless force . Three regimes can be distinguished. For values the two strands are aligned along the force direction but there is no relative motion of the last bead with respect to the first one, kept fixed in the origin, so that . In this case, the PDF of the gyration radius is narrow (see the curve corresponding to the case with and in the right panel of Figure 5). When the force is increased, the strand, which is not constrained to the origin, moves over the other part. This causes a broadening of the PDF (see the curve with ). A stronger force facilitates a larger sliding. Due to the overall fluctuations of the polymer, the final bead does not attain a fixed position relative to the first bead but can move back and forth along the chain (see the Supplementary Video S3 for and ). The PDF is characterized by more peaks corresponding to different stable configurations assumed by the polymer in the same ensemble. However, the final part of the chain cannot slide continuously due to the finite rigidity so that larger forces are required to unfold the polymer. The time behavior of the energy terms and is shown in Figure 6 in the case with for .
The last bead can slide when it occurs that , as witnessed by the increase of the end-to-end distance also reported in the figure. As becomes continuously larger, approaches and, when exceeds , the polymer swells abruptly signaling a “first-order”-like phase transition. (see the Supplementary Video S4 for and ). Once the polymer is completely elongated, the PDF has again a single peak whose position jumps discontinuously to a larger value. The force required to unzip completely the polymer increases with the bending rigidity and the transition from the folded state to the elongated one becomes sharper, as in the case of the unzipping of double-stranded DNA [39,45,69]. When the polymer is completely unfolded, the values of for different bending rigidities lay on the same curve following the decay (8) of semiflexible filaments, as it happens in the case of the stiffer chain with .
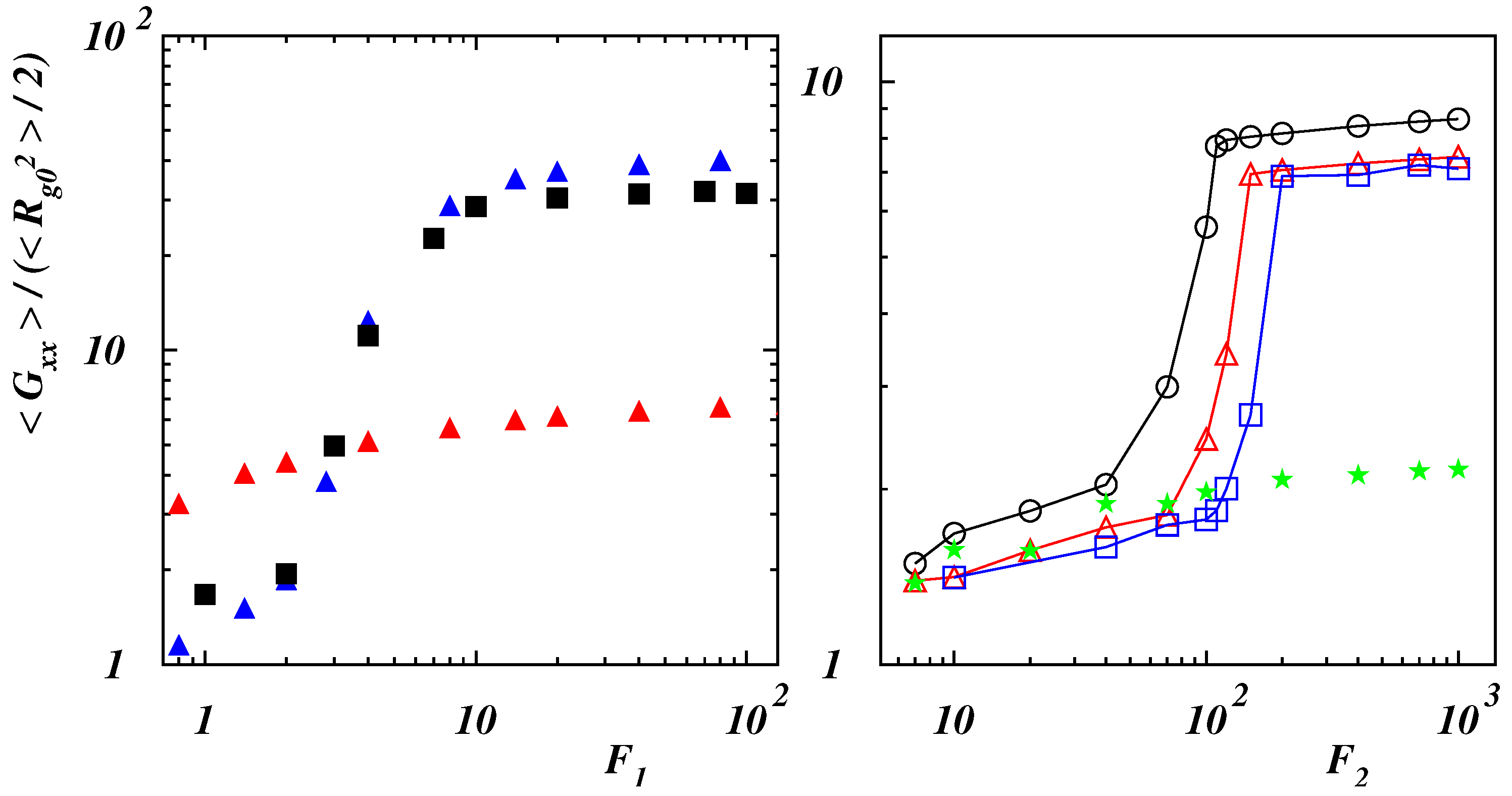
Polymer deformation can be characterized in terms of the gyration tensor (5). The ratios () are presented in Figure 7 and Figure 8, where is the mean-square value of the gyration radius calculated at equilibrium. For values of the bending rigidity (see the left panel of Figure 7), the behavior is similar and the polymer is smoothly deformed in the force direction as long as the blob is unwound ().
Once the chain has been disentangled (), the deformation reaches a value which does not change significantly with the force. Due to the initial compact structure, the ratio of deformation is considerably larger with respect to the case with negligible self-interaction which is also shown in the left panel of Figure 7 for . In the right panel of the same figure, the deformation is shown as a function of the dimensionless force when the polymer has a double-stranded initial configuration. Initially, in case of , the force slightly elongates the chain with respect to the equilibrium case. As soon as the last bead starts to slide over the filament, the deformation increases rapidly with steepness depending on the bending rigidity. Finally, it reaches a constant value when the polymer is fully elongated along the force direction. The smaller relative deformation corresponds to the more stiff polymer whose initial configuration is a stiff hairpin (see Figure 1d). When the bending rigidity is such that no closed structure can form (), the chain is smoothly elongated over the whole range of explored forces with a final value sensibly smaller than the one corresponding to initially double-stranded chains.
Along the y-direction, normal to the force, the relative deformation diminishes as a function of the dimensionless force when (left panel of Figure 8).
As long as the chain maintains a compact structure, the decrease is weak while it becomes steeper when the polymer is open under the action of the external driving. At values , data collapse and a power-law with dependence can be observed. When self-interaction is negligible, the behavior is similar but the deformation is much smaller due to the lack of a compact-like initial structure. More interesting appears to be what happens for the range of stiffness corresponding to double-stranded conformations. The initial values of decrease, due to the stretching of the two strands, with a similar trend. When the folded strands open, an overshoot can be observed that is due to the larger fluctuations of the chain. The deformation then follows a power-law decay with dependence . The data for the initially stretched polymer () show a similar behavior without the aforementioned overshoot.
4. Discussion and Conclusions
The dynamical and conformational properties of semiflexible polymers under poor-solvent condition in a uniform force field have been numerically studied. The chain has been anchored at one end, confined in two dimensions and placed in contact with a Brownian heat bath implemented by the stochastic version of the multi-particle collision dynamics.
The equilibrium conformation depends both on the stiffness and on the self-interaction strength. When the self-attraction energy is smaller compared to the thermal energy, the chain behaves as a semiflexible filament. In the opposite limit of strong mutual attraction, different configurations are obtained. At low bending rigidity the polymer assumes a compact structure. By increasing the stiffness, patterns of folded bundles emerge where the number of strands reduces as the chain becomes more rigid. A larger number of polymer beads, with respect to the value here considered, would promote folded conformations with more strands as observed for three-dimensional semiflexible polymers [70]. Finally, rod-like conformations are recovered for high values of the rigidity.
The mechanical response to the action of the external force depends on the initial equilibrium structure. For small bending rigidity the compact structure is continuously unwound and stretched by the force. On the other hand, when the polymer consists of two facing strands, a “first-order”-like phase transition is observed from the folded to the stiff conformation. These behaviors are highlighted in the force-extension relations, as well as at the probability distribution functions of the gyration radius. The deformation of the radius of gyration with respect to the equilibrium value along the direction normal to the force is found to decay as the inverse of the applied force.
Although hydrodynamics interactions have been neglected in this investigation, it is known that such interactions are not essential in the case of semiflexible polymers since only logarithmic corrections are expected [5]. Therefore, the present results also describe the behavior of a self-attractive semiflexible polymer placed in a uniform flow field as long as the chain follows the fluid flow. We hope that this study will stimulate theoretical studies and experimental investigations to confirm the outlined phenomenology.
Supplementary Materials
The following supporting information can be downloaded at: https://www.mdpi.com/article/10.3390/polym14214762/s1, Video S1: Animation of polymer stretching for and with ; Video S2: Animation of polymer stretching for and with ; Video S3: Animation of polymer stretching for and with ; Video S4: Animation of polymer stretching for and with .
Funding
This research was funded by MIUR project number PRIN 2020/PFCXPE.
Institutional Review Board Statement
Not applicable.
Data Availability Statement
Data are available upon reasonable request.
Acknowledgments
A.L. wishes to thank R. G. Winkler for critically reading the manuscript and A. Rosa for useful discussions. This work was performed under the auspices of GNFM-INdAM.
Conflicts of Interest
The author declares no conflict of interest.
References
- Shaqfeh, E.S.G. The dynamics of single-molecule DNA in flow. J. Non-Newtonian Fluid Mech. 2005, 130, 1. [Google Scholar] [CrossRef]
- Perkins, T.T.; Smith, D.E.; Larson, R.G.; Chu, S. Stretching of a single tethered polymer in a uniform flow. Science 1995, 268, 83. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wilhelm, J.; Frey, E. Radial distribution function of semiflexible polymers. Phys. Rev. Lett. 1996, 77, 2581. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Götter, R.; Kroy, K.; Frey, E.; Bärmann, M.; Sackmann, E. Dynamic Light Scattering from Semidilute Actin Solutions: A Study of Hydrodynamic Screening, Filament Bending Stiffness, and the Effect of Tropomyosin/Troponin-Binding. Macromolecules 1996, 29, 30. [Google Scholar] [CrossRef] [Green Version]
- Harnau, L.; Winkler, R.G.; Reineker, P. Dynamic Structure Factor of Semiflexible Macromolecules in Dilute Solution. J. Chem. Phys. 1996, 104, 6355. [Google Scholar] [CrossRef]
- Everaers, R.; Jülicher, F.; Ajdari, A.; Maggs, A.C. Dynamic Fluctuations of Semiflexible Filaments. Phys. Rev. Lett. 1999, 82, 3717. [Google Scholar] [CrossRef] [Green Version]
- Samuel, J.; Sinha, S. Elasticity of semiflexible polymers. Phys. Rev. E 2002, 66, 050801. [Google Scholar] [CrossRef] [Green Version]
- Le Goff, L.; Hallatschek, O.; Frey, E.; Amblard, F. Tracer Studies on F-Actin Fluctuations. Phys. Rev. Lett. 2002, 89, 258101. [Google Scholar] [CrossRef]
- Winkler, R.G. Deformation of semiflexible chains. J. Chem. Phys. 2003, 118, 2919. [Google Scholar] [CrossRef] [Green Version]
- Petrov, E.P.; Ohrt, T.; Winkler, R.G.; Schwille, P. Diffusion and Segmental Dynamics of Double-Stranded DNA. Phys. Rev. Lett. 2006, 97, 258101. [Google Scholar] [CrossRef]
- Schroeder, C.M. Single polymer dynamics for molecular rheology. J. Rheol. 2018, 62, 371. [Google Scholar] [CrossRef] [Green Version]
- Yeou, S.; Lee, N.K. Single-Molecule Methods for Investigating the Double-Stranded DNA Bendability. Mol. Cells 2022, 45, 33. [Google Scholar] [CrossRef]
- Bird, R.B.; Curtiss, C.F.; Armstrong, R.C.; Hassager, O. Dynamics of Polymer Liquids; John Wiley & Sons: New York, NY, USA, 1987; Volume 2. [Google Scholar]
- Öttinger, H.C. Stochastic Processes in Polymeric Fluids; Springer: Berlin, Germany, 1996. [Google Scholar]
- Winkler, R.G. Semiflexible polymers in shear flow. Phys. Rev. Lett. 2006, 97, 128301. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Munk, T.; Hallatschek, O.; Wiggins, C.H.; Frey, E. Dynamics of semiflexible polymers in a flow field. Phys. Rev. E 2006, 74, 041911. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Winkler, R.G. Conformational and rheological properties of semiflexible polymers in shear flow. J. Chem. Phys. 2010, 133, 164905. [Google Scholar] [CrossRef] [Green Version]
- Hur, J.S.; Shaqfeh, E.S.G. Brownian dynamics simulations of single DNA molecules in shear flow. J. Rheol. 2000, 44, 713. [Google Scholar] [CrossRef]
- Hsieh, C.C.; Larson, R.G. Modelling hydrodynamic interaction in Brownian dynamics: Simulation of extensional and shear flows of dilute solutions of high molecular weight polystyrene. J. Rheol. 2004, 48, 995. [Google Scholar] [CrossRef]
- Liu, S.; Ashok, B.; Muthukumar, M. Brownian dynamics simulations of bead-rod-chain in simple shear flow and elongational flow. Polymer 2004, 45, 1383. [Google Scholar] [CrossRef]
- Sendner, C.; Netz, R.R. Shear-induced repulsion of a semiflexible polymer from a wall. EPL 2008, 81, 54006. [Google Scholar] [CrossRef]
- He, G.L.; Messina, R.; Löwen, H.; Kiriy, A.; Bocharova, V.; Stamm, M. Shear-induced stretching of adsorbed polymer chains. Soft Matter 2009, 5, 3014. [Google Scholar] [CrossRef]
- Zhang, Y.; Donev, A.; Weisgraber, T.; Alder, B.J.; Graham, M.G.; de Pablo, J.J. Tethered DNA dynamics in shear flow. J. Chem. Phys. 2009, 130, 234902. [Google Scholar] [CrossRef] [PubMed]
- Lang, P.S.; Obermayer, B.; Frey, E. Dynamics of a semiflexible polymer or polymer ring in shear flow. Phys. Rev. E 2014, 89, 022606. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nikoubashman, A.; Howard, M.P. Equilibrium Dynamics and Shear Rheology of Semiflexible Polymers in Solution. Macromolecules 2017, 50, 8279. [Google Scholar] [CrossRef]
- Lamura, A.; Winkler, R.G. Tethered semiflexible polymer under large amplitude oscillatory shear. Polymers 2019, 11, 737. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shee, A.; Gupta, N.; Chaudhuri, A.; Chaudhuri, D. A semiflexible polymer in a gliding assay: Reentrant transition, role of turnover and activity. Soft Matter 2021, 17, 2120. [Google Scholar] [CrossRef]
- Lamura, A.; Winkler, R.G.; Gompper, G. Wall-anchored semiflexible polymer under large amplitude oscillatory shear flow. J. Chem. Phys. 2021, 154, 224901. [Google Scholar] [CrossRef]
- Andersen, N.T.; Teng, Y.; Chen, J.Z.Y. Stretching a Semiflexible Polymer of Finite Length. Macromolecules 2022, 55, 210. [Google Scholar] [CrossRef]
- Kratky, O.; Porod, G. Roentgenuntersuchung geloester Fadenmolekuele. Recl. Trav. Chim. Pays-Bas 1949, 68, 1106. [Google Scholar] [CrossRef]
- Wittmer, J.P.; Meyer, H.; Johner, A.; Kreer, T.; Baschnagel, J. Algebraic displacement correlation in two-dimensional polymer melts. Phys. Rev. Lett. 2010, 105, 037802. [Google Scholar] [CrossRef]
- Baumann, C.G.; Bloomfield, V.A.; Smith, S.B.; Bustamante, C.; Wang, M.D.; Block, S.M. Stretching of Single Collapsed DNA Molecules. Biophys. J. 2000, 78, 1965. [Google Scholar] [CrossRef]
- Gerland, U.; Bundschuh, R.; Hwa, T. Mechanically Probing the Folding Pathway of Single RNA Molecules. Biophys. J. 2003, 84, 2831. [Google Scholar] [CrossRef] [Green Version]
- Doi, M.; Edwards, S.F. The Theory of Polymer Dynamics; Clarendon Press: Oxford, UK, 1986. [Google Scholar]
- Austin, K.S.; Zierenberg, J.; Janke, W. Interplay of Adsorption and Semiflexibility: Structural Behavior of Grafted Polymers under Poor Solvent Conditions. Macromolecules 2017, 50, 4054. [Google Scholar] [CrossRef]
- Haupt, B.J.; Senden, T.J.; Sevick, E.M. AFM Evidence of Rayleigh Instability in Single Polymer Chains. Langmuir 2002, 18, 2174. [Google Scholar] [CrossRef]
- Gunari, N.; Balazs, A.C.; Walker, G.C. Force-Induced Globule-Coil Transition in Single Polystyrene Chains in Water. J. Am. Chem. Soc. 2007, 129, 10046. [Google Scholar] [CrossRef]
- Rosa, A.; Hoang, T.X.; Marenduzzo, D.; Maritan, A. Elasticity of Semiflexible Polymers with and without Self-Interactions. Macromolecules 2003, 36, 10095. [Google Scholar] [CrossRef] [Green Version]
- Tkachenko, A.V. Unfolding and unzipping of single-stranded DNA by stretching. Phys. Rev. E 2004, 70, 051901. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kumar, S.; Giri, D. Force-Induced Conformational Transition in a System of Interacting Stiff Polymers: Application to Unfolding. Phys. Rev. E 2005, 72, 052901. [Google Scholar] [CrossRef] [Green Version]
- Kneller, J.M.; Elvingson, C.; Arteca, G.A. Shape Transition Induced by Mechanical External Stretching of Grafted Self-Attractive Wormlike Chains. Chem. Phys. Lett. 2005, 407, 384. [Google Scholar] [CrossRef]
- Rosa, A.; Marenduzzo, D.; Kumar, S. Stretching a Self-Interacting Semiflexible Polymer. Europhys. Lett. 2006, 75, 818. [Google Scholar] [CrossRef] [Green Version]
- Kumar, S.; Jensen, I.; Jacobsen, J.L.; Guttmann, A.J. Role of Conformational Entropy in Force-Induced Biopolymer Unfolding. Phys. Rev. Lett. 2007, 98, 128101. [Google Scholar] [CrossRef]
- Cifra, P.; Bleha, T. Stretching of Self-Interacting Wormlike Macromolecules. Polymer 2007, 48, 2444. [Google Scholar] [CrossRef]
- Kapri, R. Can a double stranded DNA be unzipped by pulling a single strand?: Phases of adsorbed DNA. J. Chem. Phys. 2009, 130, 145105. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guttmann, A.J.; Jacobsen, J.L.; Jensen, I.; Kumar, S. Modeling Force-Induced Bio-polymer Unfolding. J. Math. Chem. 2009, 45, 223. [Google Scholar] [CrossRef] [Green Version]
- Maier, B.; Seifert, U.; Rädler, J.O. Elastic response of DNA to external electric fields in two dimensions. Europhys. Lett. 2002, 60, 622. [Google Scholar] [CrossRef]
- Kim, Y.; Kim, K.S.; Kounovsky, K.L.; Chang, R.; Jung, G.Y.; DePablo, J.J.; Kyubong, J.; Schwartz, D.C. Nanochannel confinement: DNA Stretch Approaching Full Contour Length. Lab Chip 2011, 11, 1721. [Google Scholar] [CrossRef]
- Reisner, W.; Pedersen, J.N.; Austin, R.H. DNA Confinement in Nanochannels: Physics and Biological Applications. Rep. Prog. Phys. 2012, 75, 106601. [Google Scholar] [CrossRef]
- Lamura, A.; Burkhardt, T.W.; Gompper, G. Semiflexible polymer in a uniform force field in two dimensions. Phys. Rev. E 2001, 64, 061801. [Google Scholar] [CrossRef] [Green Version]
- Lamura, A.; Winkler, R.G. Semiflexible polymers under external fields confined to two dimensions. J. Chem. Phys. 2012, 137, 244909. [Google Scholar] [CrossRef] [Green Version]
- Ripoll, M.; Winkler, R.G.; Gompper, G. Hydrodynamic screening of star polymers in shear flow. Eur. Phys. J. E 2007, 23, 349. [Google Scholar] [CrossRef]
- Kapral, R. Multiparticle Collision Dynamics: Simulations of complex systems on mesoscale. Adv. Chem. Phys. 2008, 140, 89. [Google Scholar]
- Gompper, G.; Ihle, T.; Kroll, D.M.; Winkler, R.G. Multi-Particle Collision Dynamics: A particle-based mesoscale simulation approach to the hydrodynamics of complex Fluids. Adv. Polym. Sci. 2009, 221, 1. [Google Scholar]
- Winkler, R.G.; Reineker, P.; Harnau, L. Models and equilibrium properties of stiff molecular chains. J. Chem. Phys. 1994, 101, 8119. [Google Scholar] [CrossRef]
- Hsu, H.P.; Paul, W.; Binder, K. Standard Definitions of Persistence Length Do Not Describe the Local “Intrinsic” Stiffness of Real Polymer Chains. Macromolecules 2010, 43, 3094. [Google Scholar] [CrossRef]
- Swope, W.C.; Andersen, H.C.; Berens, P.H.; Wilson, K.R. A computer simulation method for the calculation of equilibrium constants for the formation of physical clusters of molecules: Application to small water clusters. J. Chem. Phys. 1982, 76, 637. [Google Scholar] [CrossRef]
- Allen, M.P.; Tildesley, D.J. Computer Simulation of Liquids; Clarendon Press: Oxford, UK, 1987. [Google Scholar]
- Kikuchi, N.; Pooley, C.M.; Ryder, J.F.; Yeomans, J.M. Transport coefficients of a mesoscopic fluid dynamics model. J. Chem. Phys. 2003, 119, 6388–6395. [Google Scholar] [CrossRef] [Green Version]
- Ihle, T.; Kroll, D.M. Stochastic rotation dynamics: A Galilean-invariant mesoscopic model for fluid flow. Phys. Rev. E 2001, 63, 020201. [Google Scholar] [CrossRef]
- Lamura, A.; Gompper, G.; Ihle, T.; Kroll, D.M. Multiparticle collision dynamics: Flow around a circular and a square cylinder. Europhys. Lett. 2001, 56, 319–325. [Google Scholar] [CrossRef] [Green Version]
- Marko, J.F.; Siggia, E.D. Stretching DNA. Macromolecules 1995, 28, 8759. [Google Scholar] [CrossRef]
- Livadaru, L.; Netz, R.R.; Kreuzer, H.J. Stretching response of discrete semiflexible polymers. Macromolecules 2003, 36, 3732. [Google Scholar] [CrossRef]
- Rubinstein, M.; Colby, R.C. Polymer Physics; Oxford University Press: Oxford, UK, 2003. [Google Scholar]
- Dünweg, B. Polymer Solutions. In Handbook of Materials Modelling; Andreoni, W., Yip, S., Eds.; Springer: Berlin, Germany, 2018; pp. 1–19. [Google Scholar]
- Marenduzzo, D.; Maritan, A.; Rosa, A.; Seno, F. Stepwise Unfolding of Collapsed Polymers. Eur. Phys. J. E 2004, 15, 83. [Google Scholar] [CrossRef]
- Cieplak, M.; Hoang, T.X.; Robbins, M.O. Stretching of homopolymers and contact order. Phys. Rev. E 2004, 70, 011917. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lemak, A.S.; Lepock, J.R.; Chen, J.Z.Y. Molecular Dynamics Simulations of a Protein Model in Uniform and Elongational Flows. Proteins 2003, 51, 224. [Google Scholar] [CrossRef] [PubMed]
- Lubensky, D.K.; Nelson, D.R. Single molecule statistics and the polynucleotide unzipping transition. Phys. Rev. E 2002, 65, 031917. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seaton, D.T.; Schnabel, S.; Landau, D.P.; Bachmann, M. From flexible to stiff: Systematic analysis of structural phases for single semiflexible polymers. Phys. Rev. Lett. 2013, 110, 028103. [Google Scholar] [CrossRef]
Figure 1.
Equilibrium polymer conformations for (a), 0.2 (b), 0.4 (c), 0.8 (d) with . Blue and yellow beads denote the first and last ones, respectively. Polymer beads and bonds are not in scale to allow a better visualization.
Figure 1.
Equilibrium polymer conformations for (a), 0.2 (b), 0.4 (c), 0.8 (d) with . Blue and yellow beads denote the first and last ones, respectively. Polymer beads and bonds are not in scale to allow a better visualization.

Figure 2.
Root -mean-square end-to-end distance of a polymer in absence of an external force for (purple open triangles), 2 (blue filled triangles), and gyration radius for (green open circles), 2 (red filled circles). The full and dashed black lines correspond to the analytical predictions (6) and (7), respectively, in the case of continuous semiflexible polymers [55].
Figure 2.
Root -mean-square end-to-end distance of a polymer in absence of an external force for (purple open triangles), 2 (blue filled triangles), and gyration radius for (green open circles), 2 (red filled circles). The full and dashed black lines correspond to the analytical predictions (6) and (7), respectively, in the case of continuous semiflexible polymers [55].

Figure 3.
Normalized probability distribution function of the gyration radius in absence of the external force for (black open circles), (red filled circles), (green filled stars), (blue open squares), 2 (yellow open triangles) with , and for (cyan line), (purple line) with .
Figure 3.
Normalized probability distribution function of the gyration radius in absence of the external force for (black open circles), (red filled circles), (green filled stars), (blue open squares), 2 (yellow open triangles) with , and for (cyan line), (purple line) with .

Figure 4.
(Left panel) Mean deficit length-ratio along the direction of the external force as a function of the dimensionless force for (blue filled triangles), (black filled squares) with , and for (red filled triangles) with . The dashed and full lines have slopes and , respectively. (Right panel) Mean deficit length-ratio along the direction of the external force as a function of the dimensionless force for (black open circles), (red open triangles), (blue open squares), 2 (green filled stars) with . The dashed line has slope .
Figure 4.
(Left panel) Mean deficit length-ratio along the direction of the external force as a function of the dimensionless force for (blue filled triangles), (black filled squares) with , and for (red filled triangles) with . The dashed and full lines have slopes and , respectively. (Right panel) Mean deficit length-ratio along the direction of the external force as a function of the dimensionless force for (black open circles), (red open triangles), (blue open squares), 2 (green filled stars) with . The dashed line has slope .

Figure 5.
(Left panel) Normalized probability distribution function of the gyration radius for (black open circles), 4 (red filled circles), 7 (green filled stars), 20 (blue open squares) with and . (Right panel) Normalized probability distribution function of the gyration radius for (black filled squares), 120 (red filled triangles), 150 (green open triangles), 200 (blue filled stars) with and .
Figure 5.
(Left panel) Normalized probability distribution function of the gyration radius for (black open circles), 4 (red filled circles), 7 (green filled stars), 20 (blue open squares) with and . (Right panel) Normalized probability distribution function of the gyration radius for (black filled squares), 120 (red filled triangles), 150 (green open triangles), 200 (blue filled stars) with and .

Figure 6.
Potential energy differences (blue line) and (red line) as functions of time in the case of the polymer with and for . The time behavior of the end-to-end distance is also shown (black line).
Figure 6.
Potential energy differences (blue line) and (red line) as functions of time in the case of the polymer with and for . The time behavior of the end-to-end distance is also shown (black line).

Figure 7.
(Left panel) Radius of gyration tensor component along the force direction with respect to the equilibrium value as a function of the dimensionless force for (blue filled triangles), (black filled squares) with , and for (red filled triangles) with . (Right panel) Radius of gyration tensor component along the force direction with respect to the equilibrium value as a function of the dimensionless force for (black open circles), (red open triangles), (blue open squares), and 2 (green filled stars) with .
Figure 7.
(Left panel) Radius of gyration tensor component along the force direction with respect to the equilibrium value as a function of the dimensionless force for (blue filled triangles), (black filled squares) with , and for (red filled triangles) with . (Right panel) Radius of gyration tensor component along the force direction with respect to the equilibrium value as a function of the dimensionless force for (black open circles), (red open triangles), (blue open squares), and 2 (green filled stars) with .

Figure 8.
(Left panel) Radius of gyration tensor component along the y-direction with respect to the equilibrium value as a function of the dimensionless force for (blue filled triangles), (black filled squares) with , and for (red filled triangles) with . The full line has slope . (Right panel) Radius of gyration tensor component along the y-direction with respect to the equilibrium value as a function of the dimensionless force for (black open circles), (red open triangles), (blue open squares), and 2 (green filled stars) with . The dashed line has slope .
Figure 8.
(Left panel) Radius of gyration tensor component along the y-direction with respect to the equilibrium value as a function of the dimensionless force for (blue filled triangles), (black filled squares) with , and for (red filled triangles) with . The full line has slope . (Right panel) Radius of gyration tensor component along the y-direction with respect to the equilibrium value as a function of the dimensionless force for (black open circles), (red open triangles), (blue open squares), and 2 (green filled stars) with . The dashed line has slope .

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Lamura, A. Self-Attractive Semiflexible Polymers under an External Force Field. Polymers 2022, 14, 4762. https://doi.org/10.3390/polym14214762
AMA Style
Lamura A. Self-Attractive Semiflexible Polymers under an External Force Field. Polymers. 2022; 14(21):4762. https://doi.org/10.3390/polym14214762
Chicago/Turabian StyleLamura, Antonio. 2022. "Self-Attractive Semiflexible Polymers under an External Force Field" Polymers 14, no. 21: 4762. https://doi.org/10.3390/polym14214762
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.