
Effect of Hardener Type on the Photochemical and Antifungal Performance of Epoxy and Oligophosphonate S–IPNs

 ,
, 
 , , and
, , and
Abstract
:1. Introduction
2. Experimental Section
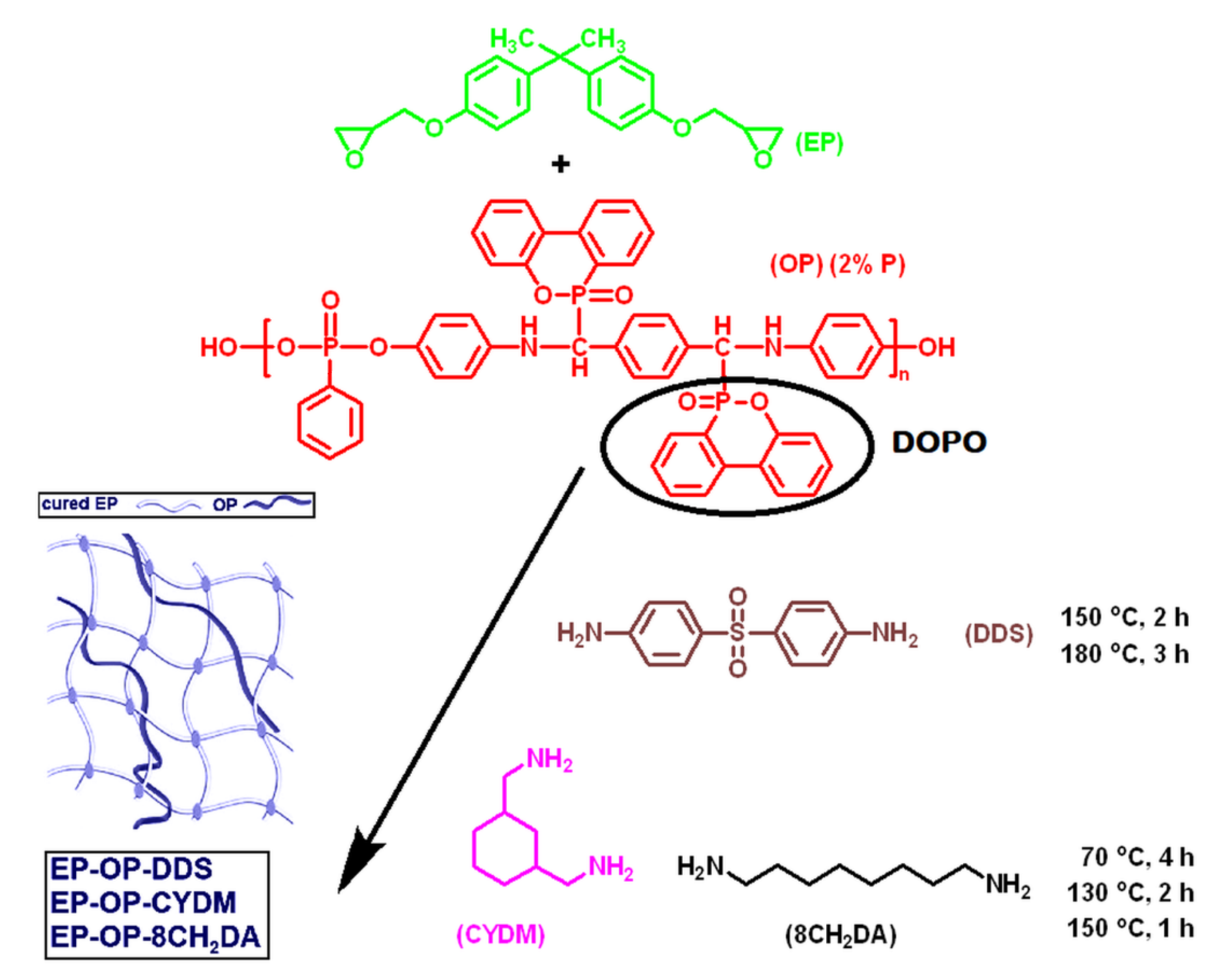
2.1. Materials
2.2. Obtaining of the S–IPNs
2.3. Measurements
2.3.1. Structural Characterization of the OP
2.3.2. Irradiation
2.3.3. Color Modification Studies
2.3.4. Structural Modifications by FT-IR during Photoirradiation
2.3.5. Morphological Study
2.3.6. Differential Scanning Calorimetry
2.3.7. Microscale Combustion Calorimetry (MCC)
2.3.8. Antifungal Behavior
3. Results and Discussions
3.1. Color Modifications during UV Irradiation
3.1.1. Lightness Factor Variation (L*)
3.1.2. Variation of Chromatic Parameter a* during UV Irradiation
3.1.3. Variation of Chromatic Parameter b* during UV Irradiation
3.1.4. Variation of Chroma Parameter C* during UV Irradiation
3.1.5. Variation of ΔE Values during UV Irradiation
3.2. Structural Modifications
FT-IR Analysis
3.3. Morphological and Thermal Characterization
3.4. Flame Resistant Capacity of the UV Irradiated S–IPNs
3.5. The Microbiological Testing of the S–IPNs
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Gurunathan, T.; Nayak, S.K. The Influence of Reactive Organoclay on a Biorenewable Castor Oil-Based Polyurethane Prepolymers Toughened Polylactide Nanocomposites. Polym. Adv. Technol. 2016, 27, 1484–1493. [Google Scholar] [CrossRef]
- Gurunathan, T.; Chung, J.S.; Nayak, S.K. Reactive Compatibilization of Biobased Polyurethane Prepolymer Toughening Polylactide Prepared by Melt Blending. J. Polym. Environ. 2016, 24, 287–297. [Google Scholar] [CrossRef]
- Cascaval, C.N.; Ciobanu, C.; Rosu, D.; Rosu, L. Polyurethane-Epoxy Maleate of Bisphenol A Semi-Interpenetrating Polymer Networks. J. Appl. Polym. Sci. 2002, 83, 138–144. [Google Scholar] [CrossRef]
- Klempner, D.; Sperling, L.H.; Utracki, L.A. Interpenetrating Polymer Netwoks; American Chemical Society: Washington, DC, USA, 1994. [Google Scholar]
- Thomas, R.; Vijayan, P.; Thomas, S. Recycling of Thermosetting Polymers. In Recent Developments in Polymer Recycling; Transworld Research Network: Trivandrum, India, 2011; pp. 122–129. [Google Scholar]
- Varganici, C.D.; Rosu, L.; Rosu, D.; Simionescu, B.C. Miscibility Studies of Some Semi–Interpenetrating Polymer Networks Based on an Aromatic Polyurethane and Epoxy Resin. Compos. Part B Eng. 2013, 50, 273–278. [Google Scholar] [CrossRef]
- Jian, R.; Wang, P.; Duan, W.; Wang, J.; Zheng, X.; Weng, J. Synthesis of a Novel P/N/S-Containing Flame Retardant and Its Application in Epoxy Resin: Thermal Property, Flame Retardance, and Pyrolysis Behavior. Ind. Eng. Chem. Res. 2016, 55, 11520–11527. [Google Scholar] [CrossRef]
- Irfan, M.H. Chemistry and Technology of Thermosetting Polymers in Construction Applications; Springer Science & Business Media: Luxembourg, 2012. [Google Scholar]
- Hayaty, M.; Honarkar, H.; Beheshty, M.H. Curing Behavior Of Dicyandiamide/Epoxy Resin System Using Different Accelerators. Iran. Polym. J. 2013, 22, 591–598. [Google Scholar] [CrossRef]
- Jin, F.L.; Park, S.J. Thermal Properties of Epoxy Resin/Filler Hybrid Composites. Polym. Degrad. Stab. 2012, 97, 2148–2153. [Google Scholar] [CrossRef]
- Skeist, I. Handbook of Adhesives; Springer Inc.: New York, NY, USA, 2011. [Google Scholar]
- Luo, C.; Zuo, J.; Zhao, J. Synthesis and Property of Epoxy Prepolymer and Curing Agent with High Refractive Index. High Perform. Polym. 2013, 25, 986–991. [Google Scholar] [CrossRef]
- Zhang, W.; Li, X.; Li, L.; Yang, R. Study of the Synergistic Effect of Silicon and Phosphorus on the Blowing–Out Effect of Epoxy Resin Composites. Polym. Degrad. Stab. 2012, 97, 1041–1048. [Google Scholar] [CrossRef]
- Toldy, A.; Szabó, A.; Novák, C.; Madarász, J.; Tóth, A.; Marosi, G. Intrinsically Flame Retardant Epoxy Resin-Fire Performance and Background–Part II. Polym. Degrad. Stab. 2008, 93, 2007–2013. [Google Scholar] [CrossRef]
- Gaan, S.; Mauclaire, L.; Rupper, P.; Salimova, V.; Tran, T.T.; Heuberger, M. Thermal Degradation of Cellulose Acetate in Presence of Bis–Phosphoramidates. J. Anal. Appl. Pyrolysis 2011, 90, 33–41. [Google Scholar] [CrossRef]
- Lin, C.H.; Chang, S.L.; Wei, T.P.; Ding, S.H.; Su, W.C. Facile, One–Pot Synthesis of Phosphinate–Substituted Bisphenol A and its Alkaline-Stable Diglycidyl Ether Derivative. Polym. Degrad. Stab. 2010, 95, 1167–1176. [Google Scholar] [CrossRef]
- Cakić, S.M.; Ristić, I.S.; Jašo, V.M.; Radičević, R.Ž.; Ilić, O.Z.; Simendić, J.K.B. Investigation of the Curing Kinetics of Alkyd–Melamine–Epoxy Resin System. Prog. Org. Coat. 2012, 73, 415–424. [Google Scholar] [CrossRef]
- Zheng, Y.; Chonung, K.; Jin, X.; Wei, P.; Jiang, P. Study on the Curing Reaction, Dielectric and Thermal Performances of Epoxy Impregnating Resin With Reactive Silicon Compounds as New Diluents. J. Appl. Polym. Sci. 2008, 107, 3127–3136. [Google Scholar] [CrossRef]
- Tseng, C.H.; Hsueh, H.B.; Chen, C.Y. Effect of Reactive Layered Double Hydroxides on the Thermal and Mechanical Properties of LDHs/Epoxy Nanocomposites. Compos. Sci. Technol. 2007, 67, 2350–2362. [Google Scholar] [CrossRef]
- Shi, Y.Q.; Fu, T.; Xu, Y.J.; Li, D.F.; Wang, X.L.; Wang, Y.Z. Novel Phosphorus-Containing Halogen-Free Ionic Liquid Toward Fire Safety Epoxy Resin with Well-Balanced Comprehensive Performance. Chem. Eng. J. 2018, 354, 208–219. [Google Scholar] [CrossRef]
- Kou, Y.J.; Zhou, W.Y.; Li, B.; Dong, L.; Duan, Y.E.; Hou, Q.W.; Liu, X.R.; Cai, H.W.; Chen, Q.G.; Dang, Z.M. Enhanced Mechanical and Dielectric Properties of an Epoxy Resin Modified With Hydroxyl-Terminated Polybutadiene. Compos. Part A Appl. Sci. 2018, 114, 97–106. [Google Scholar] [CrossRef]
- Laurien, M.; Demir, B.; Buttemeyer, H.; Herrmann, A.S.; Walsh, T.R.; Ciacchi, L.C. Atomistic Modeling of the Formation of a Thermoset/Thermoplastic Interphase During Co-curing. Macromolecules 2018, 51, 3983–3993. [Google Scholar] [CrossRef]
- Zhao, M.; Meng, L.H.; Ma, L.C.; Ma, L.N.; Yang, X.B.; Huang, Y.D.; Ryu, J.E.; Shankar, A.; Li, T.X.; Yan, C.; et al. Layer–by–Layer Grafting CNTs Onto Carbon Fibers Surface for Enhancing the Interfacial Properties of Epoxy Resin Composites. Compos. Sci. Technol. 2018, 154, 28–36. [Google Scholar] [CrossRef]
- Salmeia, K.A.; Gaan, S. An Overview of Some Recent Advances in DOPO–derivatives: Chemistry and Flame Retardant Applications. Polym. Degrad. Stab. 2015, 113, 119–134. [Google Scholar] [CrossRef]
- Roy, S.; Maiti, S. Synthesis and Characterization of Thermally Stable Cardo Polyphosphonates. Polymer 1998, 39, 3809–3813. [Google Scholar] [CrossRef]
- Varganici, C.D.; Rosu, L.; Bifulco, A.; Rosu, D.; Mustata, F.; Gaan, S. Recent Advances in Flame Retardant Epoxy Systems From Reactive DOPO-Based Phosphorus Additives. Polym. Degrad. Stab. 2022, 202, 110020. [Google Scholar] [CrossRef]
- Bifulco, A.; Parida, D.; Salmeia, K.A.; Nazir, R.; Lehner, S.; Stämpfli, R.; Markus, H.; Malucelli, G.; Branda, F.; Gaan, S. Fire and Mechanical Properties of DGEBA-Based Epoxy Resin Cured With a Cycloaliphatic Hardener: Combined Action of Silica, Melamine and DOPO-Derivative. Mater. Des. 2020, 193, 108862. [Google Scholar] [CrossRef]
- Carja, I.D.; Serbezeanu, D.; Vlad-Bubulac, T.; Hamciuc, C.; Coroaba, A.; Lisa, G.; López, C.G.; Soriano, M.F.; Pérez, V.F.; Sánchez, M.D.R. A Straightforward, Eco-Friendly and Cost-Effective Approach Towards Flame Retardant Epoxy Resins. J. Mater. Chem. A 2014, 2, 16230–16241. [Google Scholar] [CrossRef]
- Chen, L.; Wang, Y.Z. Aryl Polyphosphonates: Useful Halogen-Free Flame Retardants for Polymers. Materials 2010, 3, 4746–4760. [Google Scholar] [CrossRef]
- Varganici, C.D.; Rosu, L.; Lehner, S.; Hamciuc, C.; Jovic, M.; Rosu, D.; Mustata, F.; Gaan, S. Semi–Interpenetrating Networks Based on Epoxy Resin and Oligophosphonate: Comparative Effect of Three Hardeners on the Thermal and Fire Properties. Mater. Des. 2021, 212, 110237. [Google Scholar] [CrossRef]
- Reinprecht, L.; Hulla, M. Colour Changes in Beech Wood Modified With Essential Oils Due to Fungal and Ageing-Fungal Attacks with Coniophora Puteana. Drewno 2015, 58, 37–48. [Google Scholar] [CrossRef]
- Cogulet, A.; Blanchet, P.; Landry, V. Wood Degradation Under UV Irradiation: A Lignin Characterization. J. Photochem. Photobiol. B Biol. 2016, 158, 184–191. [Google Scholar] [CrossRef]
- Gu, X.; Stanley, D.; Byrd, W.E.; Dickens, B.; Vaca-Trigo, I.; Meeker, W.Q.; Nguyen, T.; Chin, J.W.; Martin, J.W. Linking Accelerated Laboratory Test with Outdoor Performance Results for a Model Epoxy Coating System. In Service Life Prediction of Polymeric Materials—Global Perspectives; Springer Science and Business Media: New York, NY, USA, 2009; pp. 3–28. [Google Scholar]
- Rosu, D.; Rosu, L.; Cascaval, C.N. Effect of Ultraviolet Radiation on Vinyl Ester Network Based on Bisphenol A. J. Photochem. Photobiol. A Chem. 2008, 194, 275–282. [Google Scholar] [CrossRef]
- Larché, J.F.; Bussière, P.O.; Thérias, S.; Gardette, J.L. Photooxidation of Polymers: Relating Material Properties to Chemical Changes. Polym. Degrad. Stab. 2012, 97, 25–34. [Google Scholar] [CrossRef]
- Silverstein, R.M.; Webster, F.S.; Kiemle, D.J. Spectrometric Identification of Organic Compounds; John Wiley & Sons: Hoboken, NJ, USA, 2005. [Google Scholar]
- Field, L.D.; Li, H.L.; Magill, A.M. Organic Structures from Spectra, 6th ed.; Wiley: Hoboken, NJ, USA, 2020. [Google Scholar]
- Delor-Jestin, F.; Drouin, D.; Cheval, P.Y.; Lacoste, J. Thermal and Photochemical Ageing of Epoxy Resin—Influence of Curing Agents. Polym. Degrad. Stab. 2006, 91, 1247–1255. [Google Scholar] [CrossRef]
- Vera-Graziano, R.; Hernandez-Sanchez, F.; Cauich-Rodriguez, J.V. Study of Crosslinking Density in Polydimethylsiloxane Networks by DSC. J. Appl. Polym. Sci. 1995, 55, 1317–1327. [Google Scholar] [CrossRef]

















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| S–IPN | Hardener (g) | OP (g) | EP (g) | |
|---|---|---|---|---|
| EP–OP–DDS | DDS | 5.99 | 5.61 | 18.36 |
| EP–OP–CYDM | CYDM | 3.79 | 5.56 | 20.63 |
| EP–OP–8CH2DA | 8CH2DA | 3.87 | 5.65 | 20.50 |
| Sample | ΔCp (J g−1 °C) | ρc’ (mol cm−3) |
|---|---|---|
| OP | 0.314 | – |
| EP–OP–DDS | 0.352 | 0.121 |
| EP–OP–CYDM | 0.335 | 0.066 |
| EP–OP–8CH2DA | 0.321 | 0.023 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Varganici, C.-D.; Rosu, L.; Rosu, D.; Hamciuc, C.; Rosca, I.; Vasiliu, A.-L. Effect of Hardener Type on the Photochemical and Antifungal Performance of Epoxy and Oligophosphonate S–IPNs. Polymers 2022, 14, 3784. https://doi.org/10.3390/polym14183784
Varganici C-D, Rosu L, Rosu D, Hamciuc C, Rosca I, Vasiliu A-L. Effect of Hardener Type on the Photochemical and Antifungal Performance of Epoxy and Oligophosphonate S–IPNs. Polymers. 2022; 14(18):3784. https://doi.org/10.3390/polym14183784
Chicago/Turabian StyleVarganici, Cristian-Dragos, Liliana Rosu, Dan Rosu, Corneliu Hamciuc, Irina Rosca, and Ana-Lavinia Vasiliu. 2022. "Effect of Hardener Type on the Photochemical and Antifungal Performance of Epoxy and Oligophosphonate S–IPNs" Polymers 14, no. 18: 3784. https://doi.org/10.3390/polym14183784