
Improving Mechanical Properties of Starch-Based Hydrogels Using Double Network Strategy
 and
and
Abstract
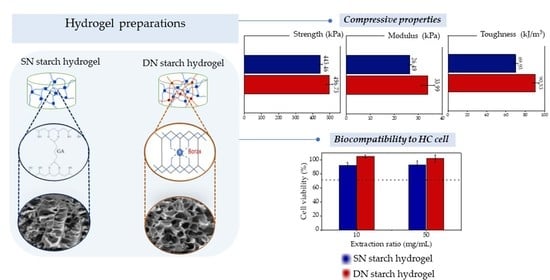
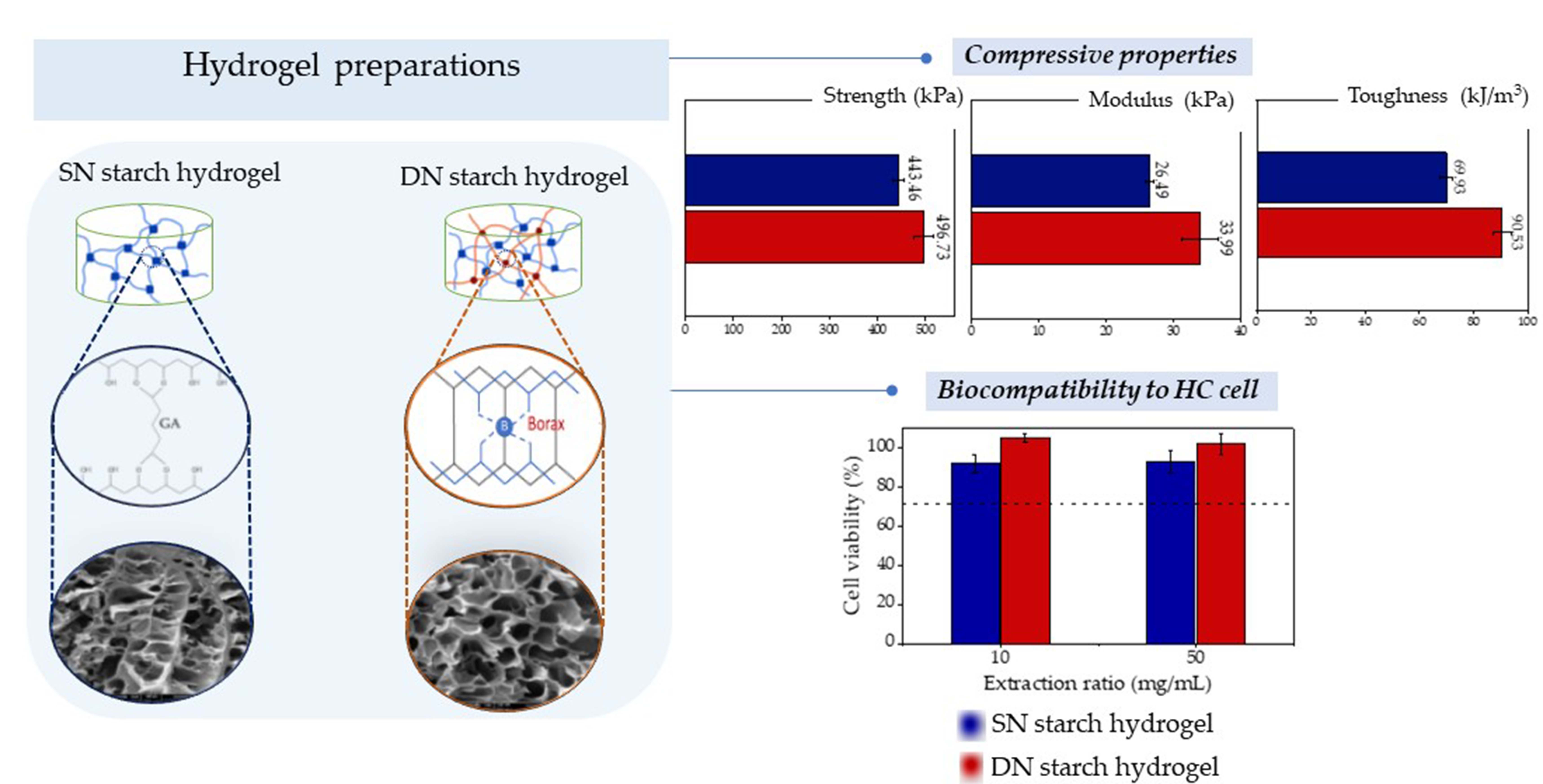
:
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Methods
2.2.1. Preparations
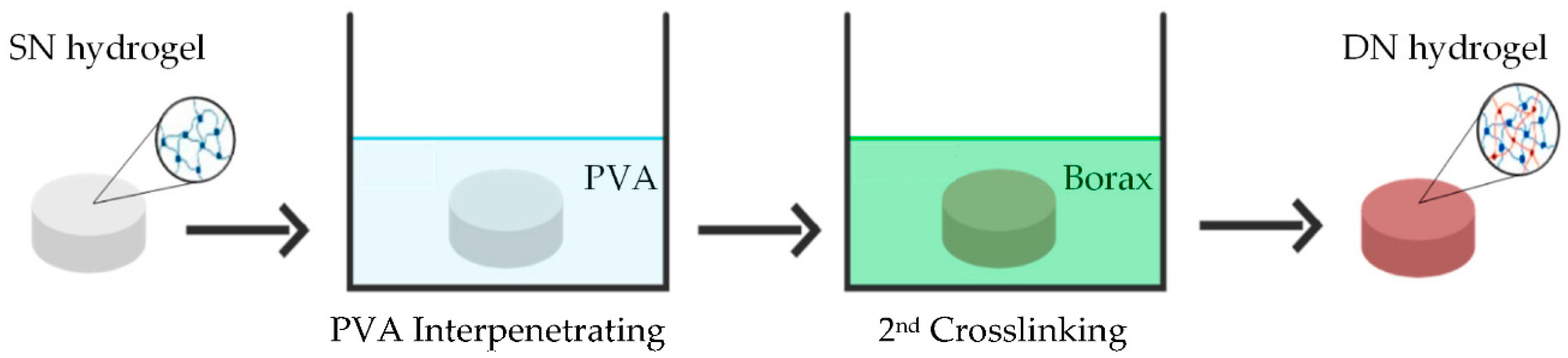
- Preparation of single network (SN) starch hydrogels
- Preparation of double network (DN) starch hydrogels
2.2.2. Chemical Structure Analysis
2.2.3. Morphology
2.2.4. Porosity
2.2.5. Water Uptake Capacity
2.2.6. Compressive Properties
2.2.7. In Vitro Cytotoxicity
2.3. Statistical Analysis
3. Results and Discussion
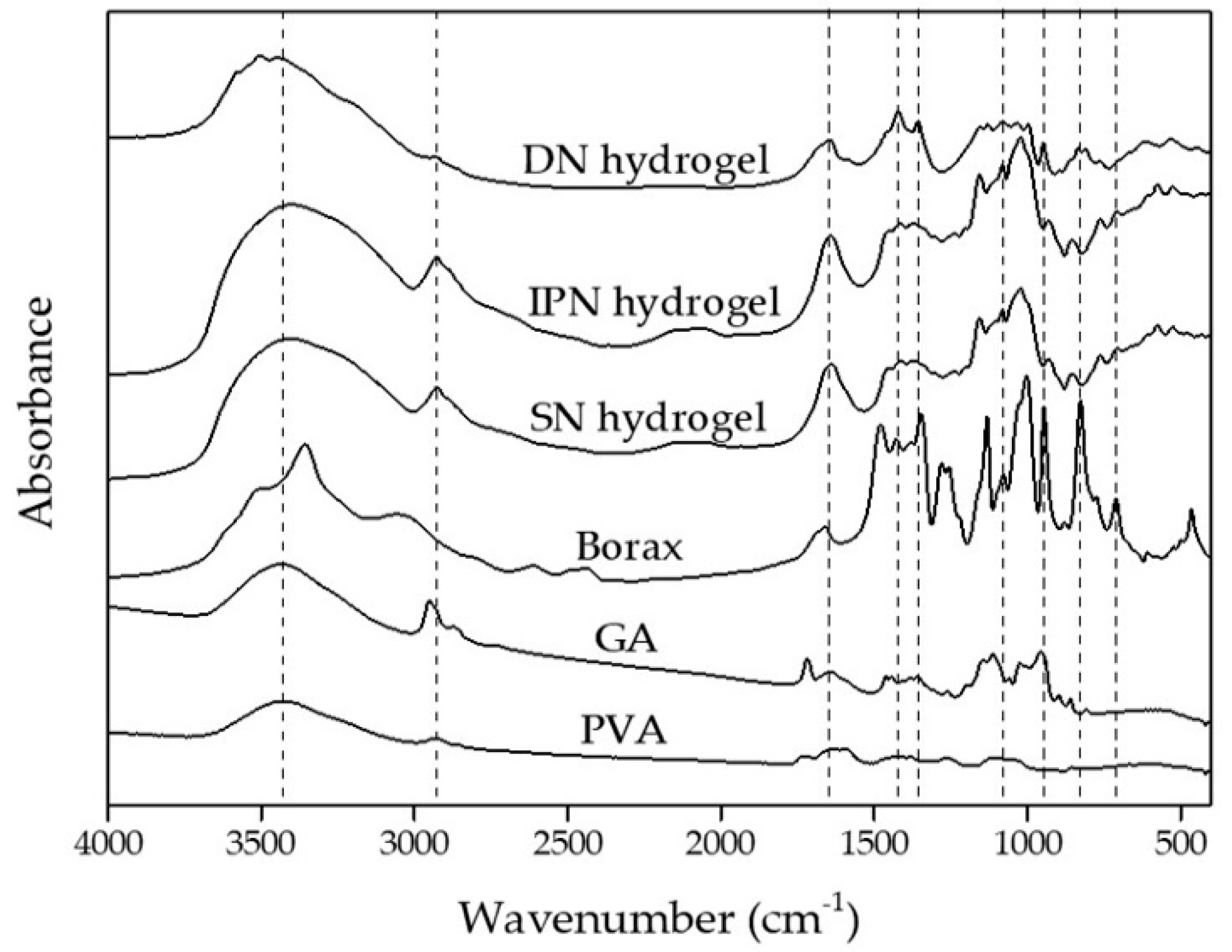
3.1. Chemical Structure Analysis of Starch Hydrogels
3.2. Characterization of Single Network (SN) Starch Hydrogels
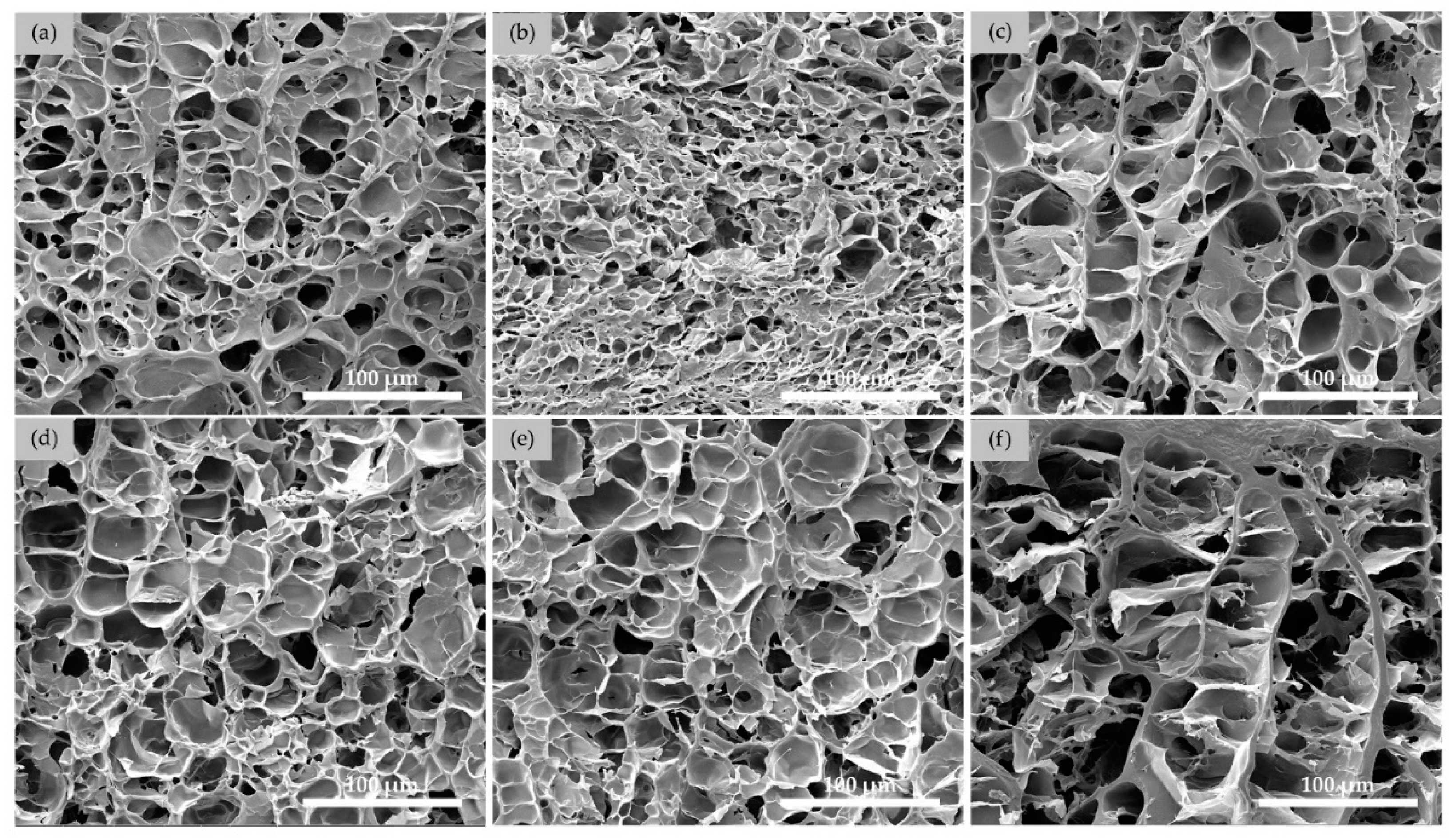
3.2.1. Morphologies of SN Starch Hydrogels
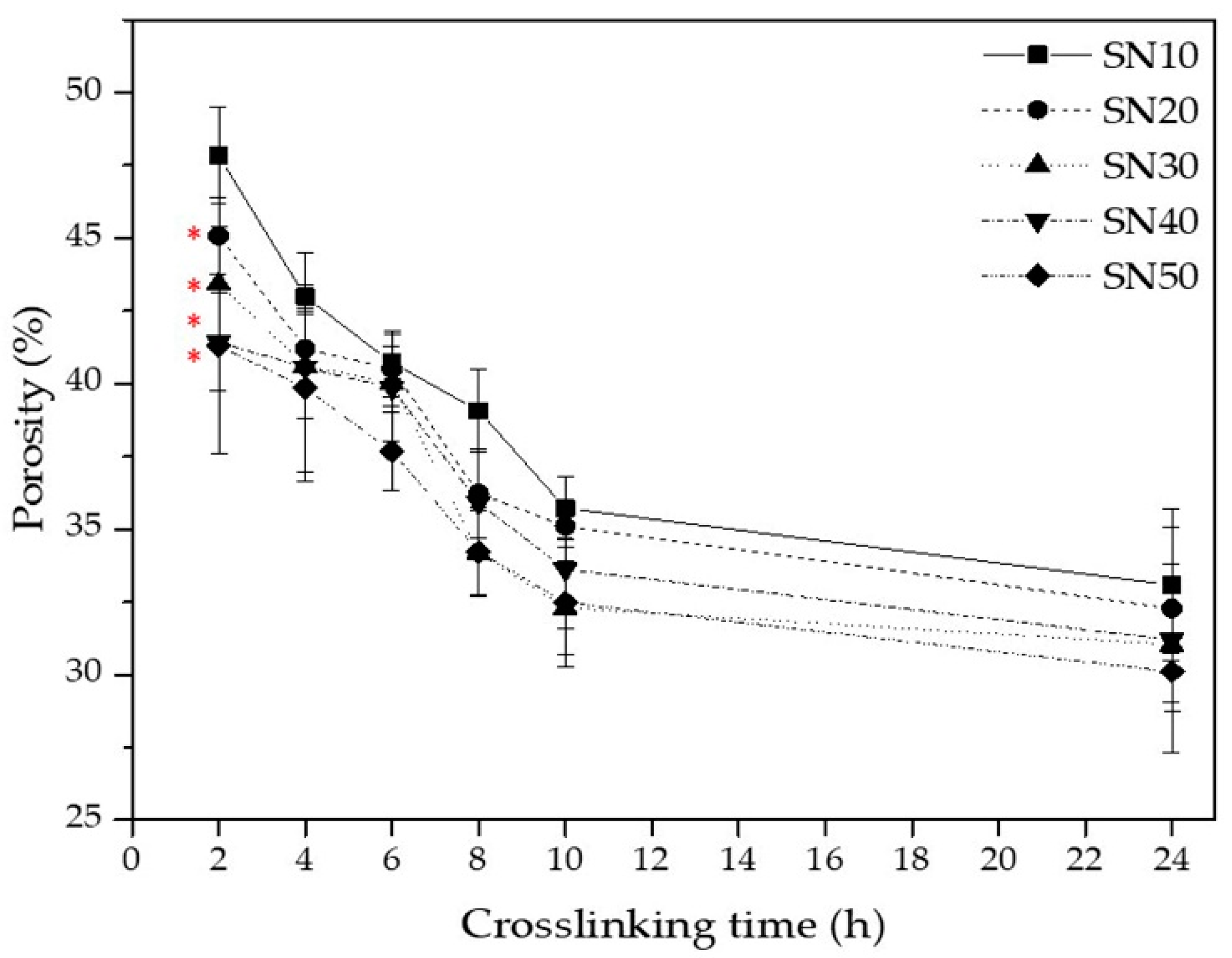
3.2.2. Porosity of SN Starch Hydrogels
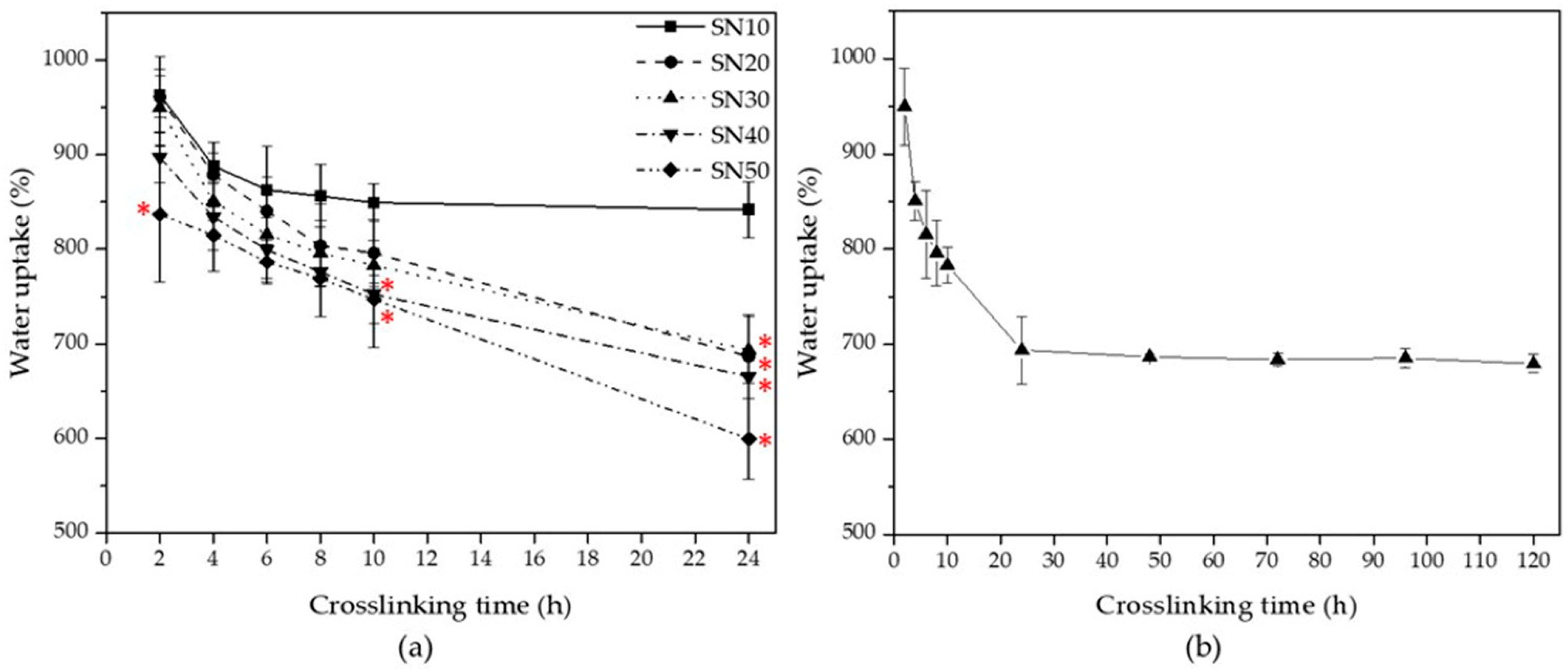
3.2.3. Water Uptake of SN Starch Hydrogels
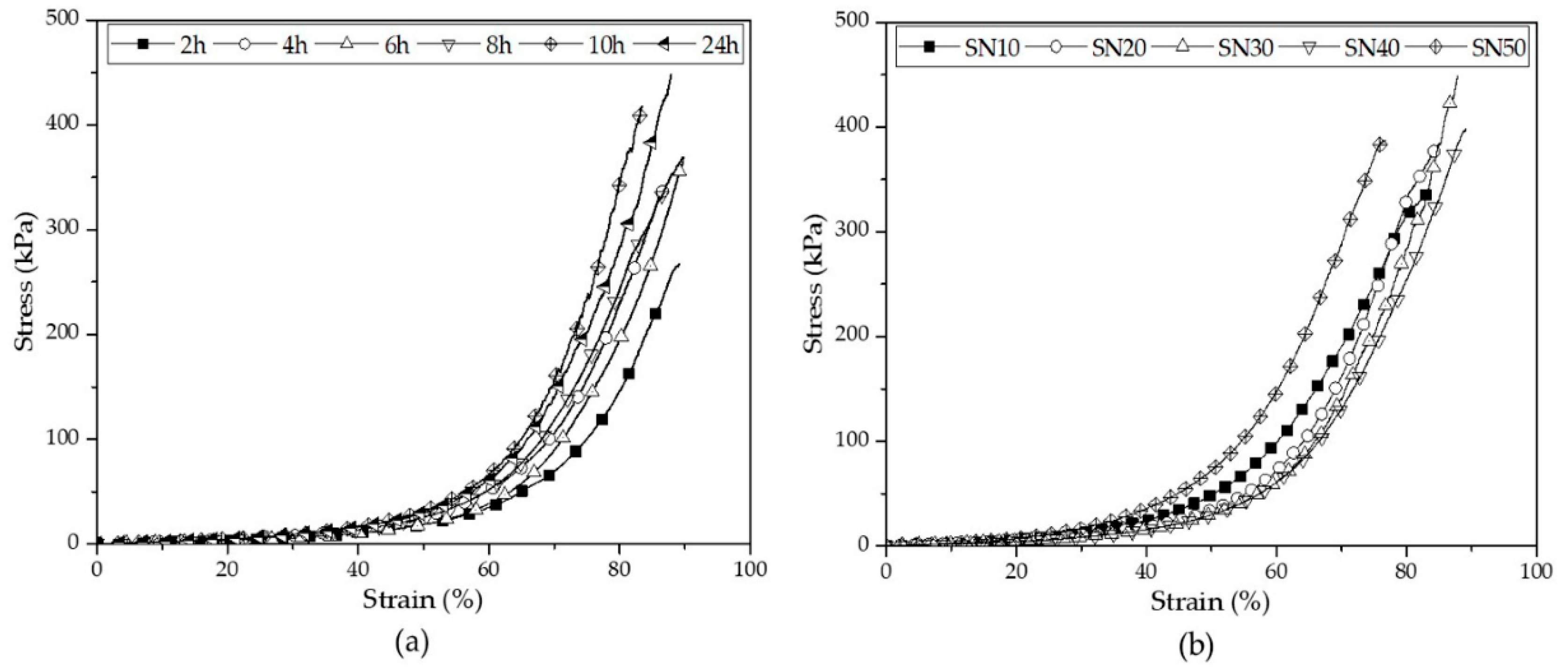
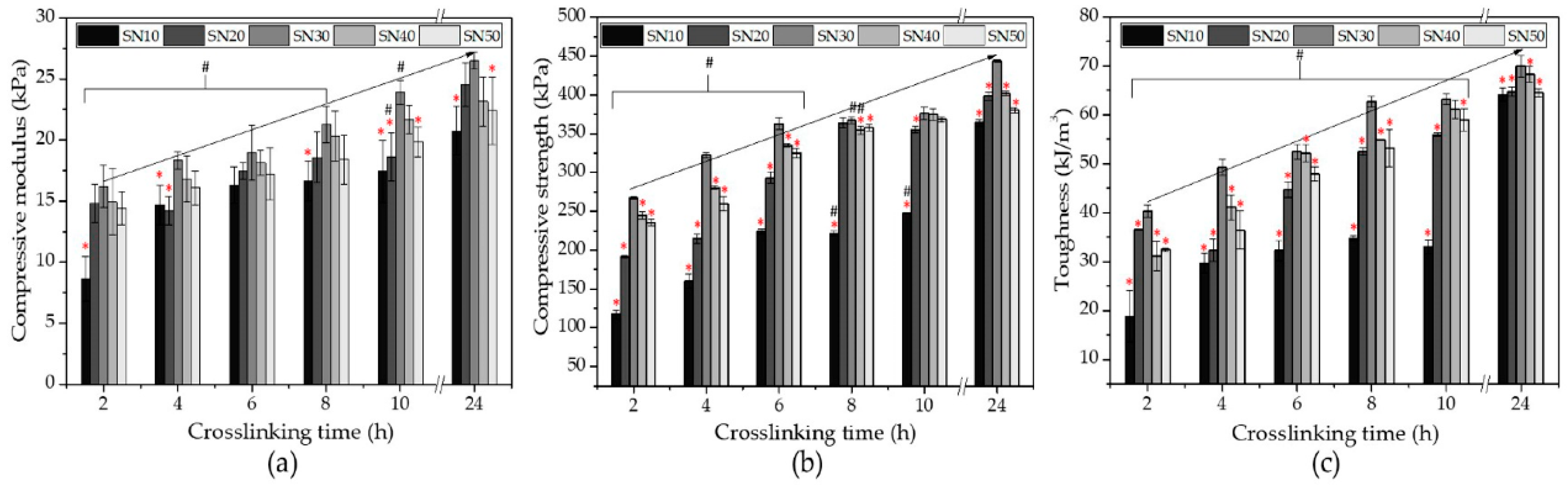
3.2.4. Compressive Properties of SN Starch Hydrogels
3.3. Characterization of Double Network (DN) Starch Hydrogels
3.3.1. Morphologies of DN Hydrogels
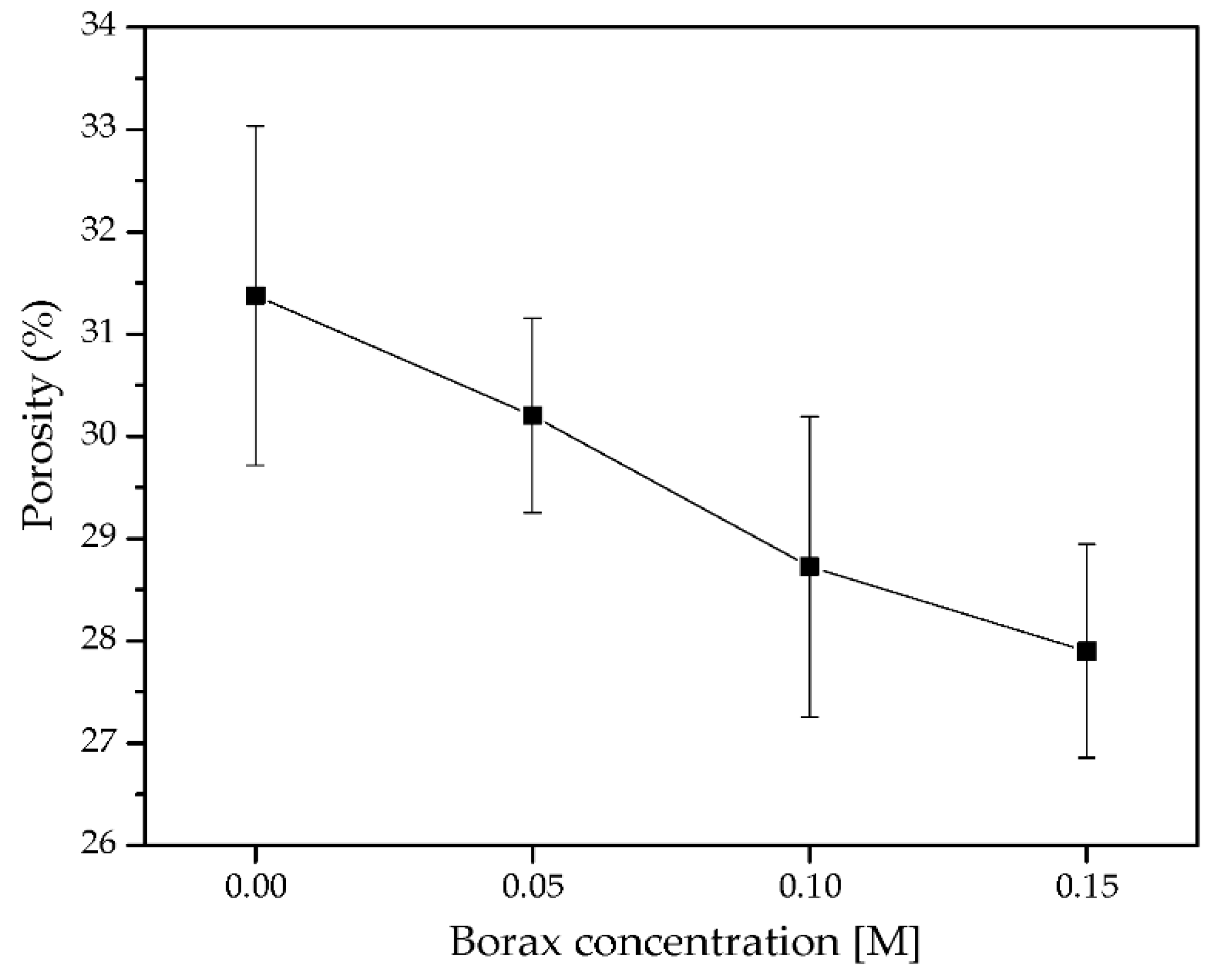
3.3.2. Porosity of DN Starch Hydrogels
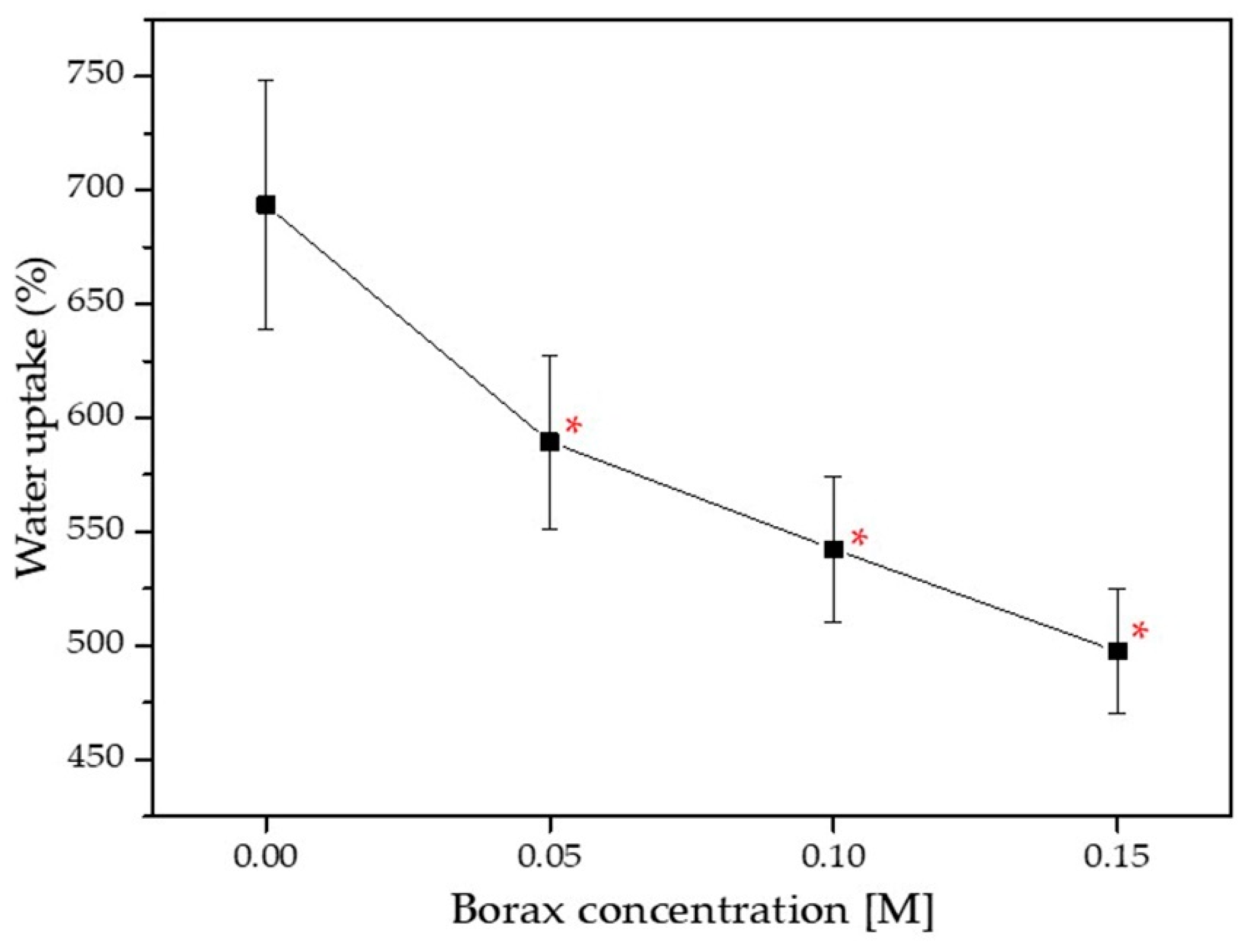
3.3.3. Water Uptake of DN Starch Hydrogels
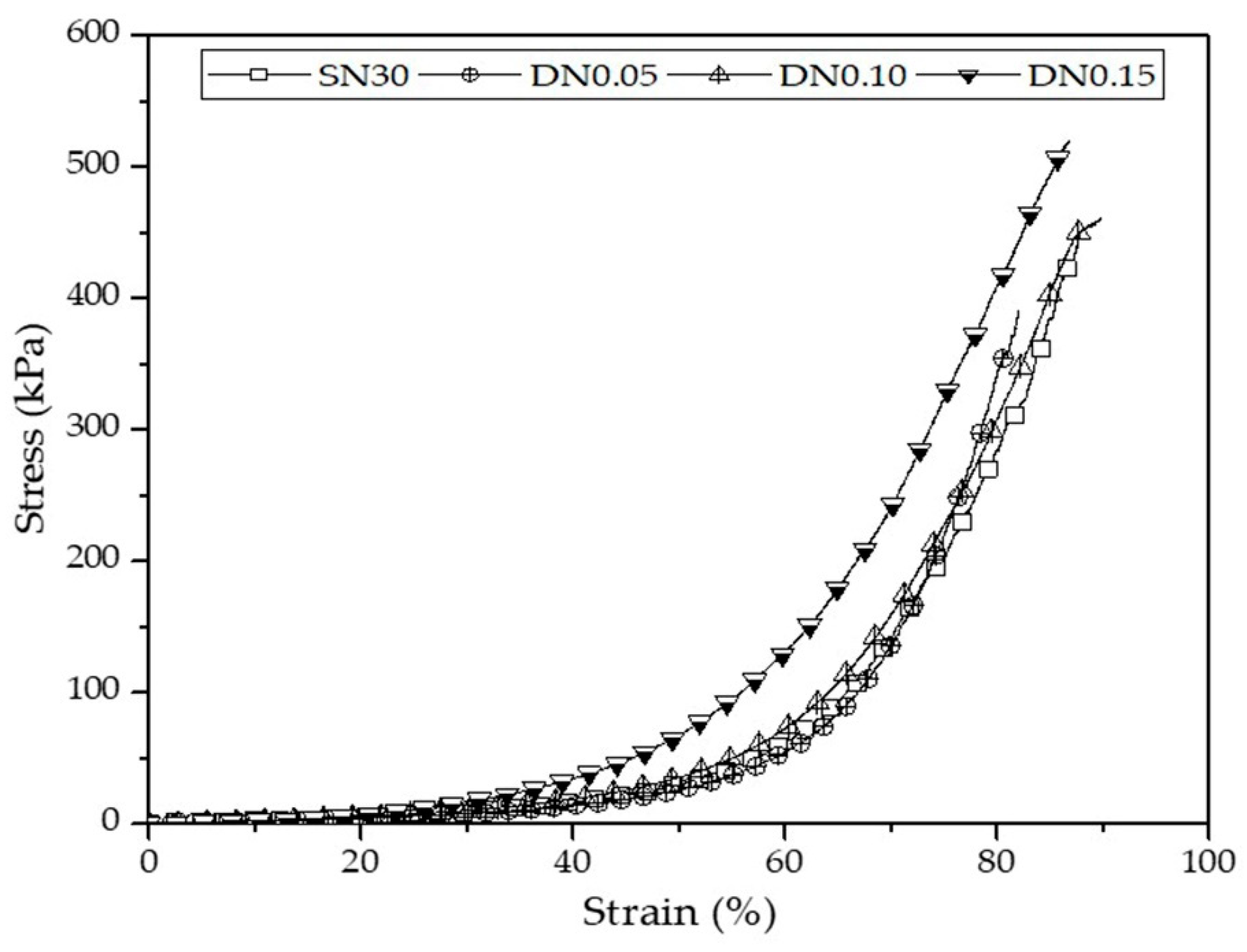
3.3.4. Compressive Properties of DN Starch Hydrogels
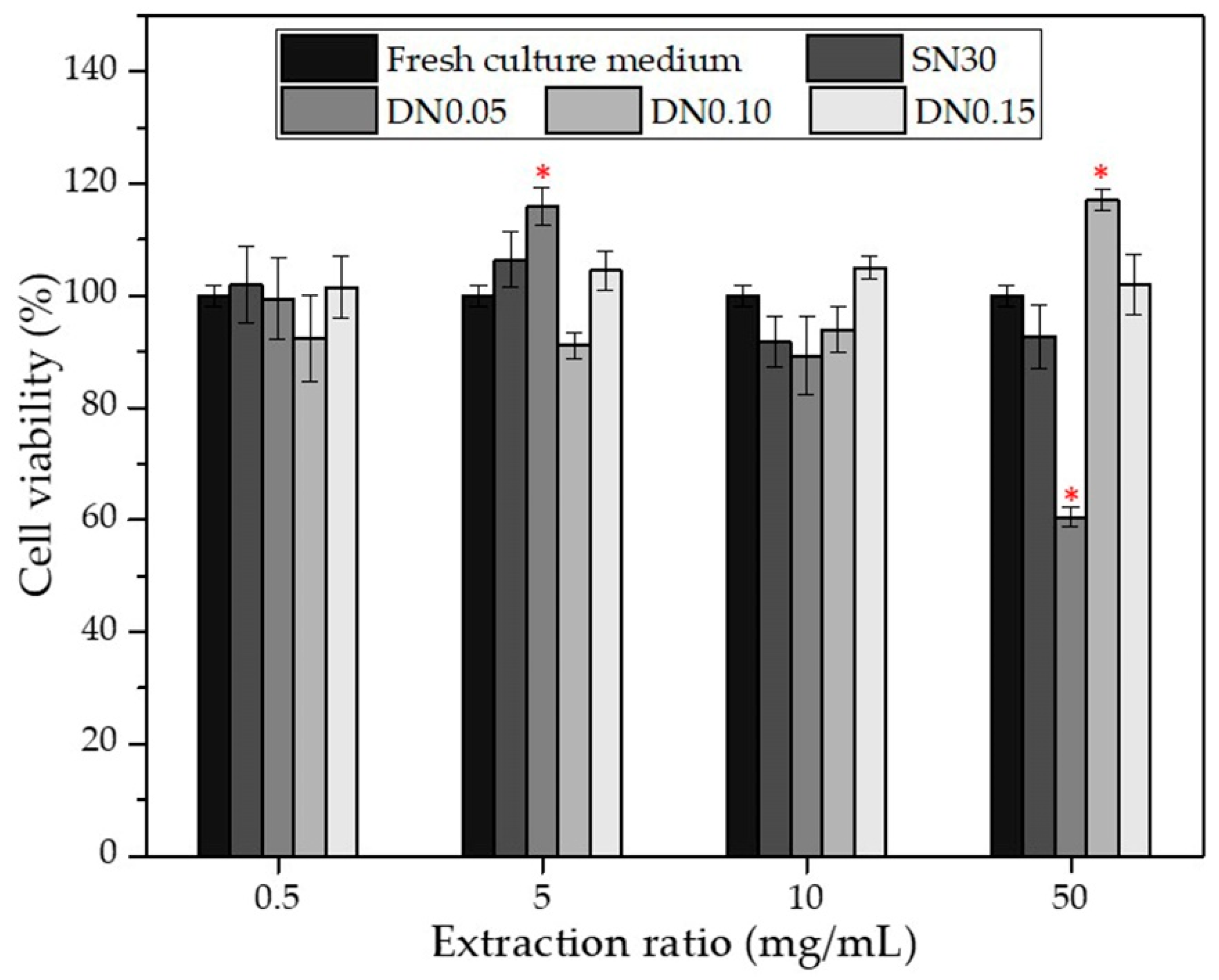
3.4. In Vitro Cytotoxicity
4. Conclusions
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Mirab, F.; Eslamian, M.; Bagheri, R. Fabrication and Characterization of a Starch-Based Nanocomposite Scaffold with Highly Porous and Gradient Structure for Bone Tissue Engineering. Biomed. Phys. Eng. Express 2018, 4, 055021. [Google Scholar] [CrossRef]
- Biliaderis, C.G. The Structure and Interactions of Starch with Food Constituents. Can. J. Physiol. Pharmacol. 1991, 69, 60–78. [Google Scholar] [CrossRef] [PubMed]
- Ismail, H.; Irani, M.; Ahmad, Z. Starch-Based Hydrogels: Present Status and Applications. Int. J. Polym. Mater. Polym. Biomater. 2013, 62, 411–420. [Google Scholar] [CrossRef]
- Qamruzzaman, M.; Ahmed, F.; Mondal, M.I.H. An Overview on Starch-Based Sustainable Hydrogels: Potential Applications and Aspects. J. Polym. Environ. 2022, 30, 19–50. [Google Scholar] [CrossRef]
- Garg, S.; Garg, A. Hydrogel: Classification, Properties, Preparation and Technical Features. Asian J. Biomater. Res. 2016, 2, 163–170. [Google Scholar]
- El-Sherbiny, I.M.; Yacoub, M.H. Hydrogel scaffolds for tissue engineering: Progress and challenges. Glob. Cardiol. Sci. Pract. 2013, 3, 316–342. [Google Scholar] [CrossRef]
- Drury, J.L.; Mooney, D.J. Hydrogels for tissue engineering: Scaffold design variables and applications. Biomaterials 2003, 24, 4337–4351. [Google Scholar] [CrossRef]
- Qi, C.; Liu, J.; Jin, Y.; Xu, L.; Wang, G.; Wang, Z.; Wang, L. Photo-crosslinkable, injectable sericin hydrogel as 3D biomimetic extracellular matrix for minimally invasive repairing cartilage. Biomaterials 2018, 163, 89–104. [Google Scholar] [CrossRef]
- Geckil, H.; Xu, F.; Zhang, X.; Moon, S.; Demirci, U. Engineering hydrogels as extracellular matrix mimics. Nanomedicine 2010, 5, 469–484. [Google Scholar] [CrossRef]
- Anwar, R.; Hughes, A.; Hama, J.; Wang, W.; Tai, H. Hydrogels from Dextran and Soybean Oil by UV Photo-Polymerization. J. Appl. Polym. Sci. 2015, 132, 41446. [Google Scholar]
- Gadhave, R.V.; Mahanwar, P.A.; Gadekar, P.T. Effect of glutaraldehyde on thermal and mechanical properties of starch and poly(vinyl alcohol) blends. Des. Monomers Polym. 2019, 22, 164–170. [Google Scholar] [CrossRef] [PubMed]
- Dixit, A.; Bag, D.S.; Kalra, S.J.S. Synthesis of strong and stretchable double network (DN) hydrogels of PVA-borax and P(AM-HEMA) and study of their swelling kinetics and mechanical properties. Polymer 2017, 119, 263–273. [Google Scholar] [CrossRef]
- Bao, Z.; Xian, C.; Yuan, Q.; Liu, G.; Wu, J. Natural Polymer-Based Hydrogels with Enhanced Mechanical Performances: Preparation, Structure, and Property. Adv. Healthc. Mater. 2019, 8, 1900670. [Google Scholar] [CrossRef]
- Panyamao, P.; Ruksiriwanich, W.; Sirisa-Ard, P.; Charumanee, S. Injectable Thermosensitive Chitosan/Pullulan-Based Hydrogels with Improved Mechanical Properties and Swelling Capacity. Polymers 2020, 12, 2514. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; He, W.; Zhang, Z.; Lee, B.P. Recent Developments in Tough Hydrogels for Biomedical Applications. Gels 2018, 4, 46. [Google Scholar] [CrossRef]
- Minhas, M.; Ahmad, M.; Ali, D.L.; Sohail, M. Synthesis of chemically cross-linked poly(vinyl alcohol)-co-poly (methacrylic acid) hydrogels by copolymerization; a potential graft-polymeric carrier for oral delivery of 5-fluorouracil. DARU J. Pharm. Sci. 2013, 21, 44. [Google Scholar] [CrossRef]
- Kumeta, K.; Nagashima, I.; Matsui, S.; Mizoguchi, K. Crosslinking of Poly(vinyl alcohol) via Bis(β-hydroxyethyl) Sulfone. Polym. J. 2004, 36, 472–477. [Google Scholar] [CrossRef]
- Lu, B.; Lin, F.; Jiang, X.; Cheng, J.; Lu, Q.; Song, J.; Chen, C.; Huang, B. One-Pot Assembly of Microfibrillated Cellulose Reinforced PVA–Borax Hydrogels with Self-Healing and pH-Responsive Properties. ACS Sustain. Chem. Eng. 2017, 5, 948–956. [Google Scholar] [CrossRef]
- Paradossi, G.; Cavalieri, F.; Chiessi, E.; Spagnoli, C.; Cowman, M. Poly(vinyl alcohol) as versatile biomaterial for potential biomedical applications. J. Mater. Sci. Mater. Med. 2003, 14, 687–691. [Google Scholar] [CrossRef]
- Stammen, J.A.; Williams, S.; Ku, D.N.; Guldberg, R.E. Mechanical properties of a novel PVA hydrogel in shear and unconfined compression. Biomaterials 2001, 22, 799–806. [Google Scholar] [CrossRef]
- Yarimitsu, S.; Sasaki, S.; Murakami, T.; Suzuki, A. Evaluation of lubrication properties of hydrogel artificial cartilage materials for joint prosthesis. Biosurf. Biotribol. 2016, 2, 40–47. [Google Scholar]
- Ngadimin, K.D.; Stokes, A.; Gentile, P.; Ferreira, A.M. Biomimetic hydrogels designed for cartilage tissue engineering. Biomater. Sci. 2021, 9, 4246–4259. [Google Scholar] [CrossRef] [PubMed]
- Yu, F.; Yang, P.; Yang, Z.; Zhang, X.; Ma, J. Double-network hydrogel adsorbents for environmental applications. Chem. Eng. J. 2021, 426, 131900. [Google Scholar] [CrossRef]
- Tian, Z.; Liu, W.; Li, G. The microstructure and stability of collagen hydrogel cross-linked by glutaraldehyde. Polym. Degrad. Stabil. 2016, 130, 264–270. [Google Scholar] [CrossRef]
- Mansur, H.S.; Oréfice, R.L.; Mansur, A.A.P. Characterization of poly(vinyl alcohol)/poly(ethylene glycol) hydrogels and PVA-derived hybrids by small-angle X-ray scattering and FTIR spectroscopy. Polymer 2004, 45, 7193–7202. [Google Scholar]
- Ramachandran, S.; Sathyamoorthy, N.; Dhanaraju, M.D. Formulation and Characterization of Glutaraldehyde Cross-Linked Chitosan Biodegradable Microspheres Loaded with Famotidine. Trop. J. Pharm. Res. 2011, 10, 309–316. [Google Scholar] [CrossRef]
- Podhorská, B.; Vetrík, M.; Chylíková-Krumbholcová, E.; Kománková, L.; Rashedi Banafshehvaragh, N.; Šlouf, M.; Dušková-Smrčková, M.; Janoušková, O. Revealing the True Morphological Structure of Macroporous Soft Hydrogels for Tissue Engineering. Appl. Sci. 2020, 10, 6672. [Google Scholar] [CrossRef]
- Shirbin, S.; Karimi, F.; Chan, N.; Heath, D.; Qiao, G. Macroporous Hydrogels Composed Entirely of Synthetic Polypeptides: Biocompatible and Enzyme Biodegradable 3D Cellular Scaffolds. Biomacromolecules 2016, 17, 2981–2991. [Google Scholar] [CrossRef]
- Llorens-Gámez, M.; Salesa, B.; Serrano-Aroca, Á. Physical and biological properties of alginate/carbon nanofibers hydrogel films. Int. J. Biol. Macromol. 2020, 151, 499–507. [Google Scholar] [CrossRef]
- Pankongadisak, P.; Suwantong, O. Enhanced properties of injectable chitosan-based thermogelling hydrogels by silk fibroin and longan seed extract for bone tissue engineering. Int. J. Biol. Macromol. 2019, 138, 412–424. [Google Scholar]
- Abdullah, A.H.D.; Chalimah, S.; Primadona, I.; Hanantyo, M.H.G. Physical and chemical properties of corn, cassava, and potato starches. IOP Conf. Ser. Earth Environ. Sci. 2018, 160, 012003. [Google Scholar] [CrossRef]
- Jain, S.; Chattopadhyay, S.; Jackeray, R.; Singh, H. Surface modification of polyacrylonitrile fiber for immobilization of anti-bodies and detection of analyte. Anal. Chim. Acta 2009, 654, 103–110. [Google Scholar] [PubMed]
- Omer, R.A.; Hama, J.R.; Rashid, R.S.M. The Effect of Dextran Molecular Weight on the Biodegradable Hydrogel with Oil, Synthesized by the Michael Addition Reaction. Adv. Polym. Technol. 2017, 36, 120–127. [Google Scholar]
- Mansur, H.S.; Sadahira, C.M.; Souza, A.N.; Mansur, A.A.P. FTIR spectroscopy characterization of poly(vinyl alcohol) hydrogel with different hydrolysis degree and chemically crosslinked with glutaraldehyde. Mater. Sci. Eng. C 2008, 28, 539–548. [Google Scholar]
- Goel, N.; Sinha, N.; Kumar, B. Growth and properties of sodium tetraborate decahydrate single crystals. Mater. Res. Bull. 2013, 48, 1632–1636. [Google Scholar]
- Bi, L.; Cao, Z.; Hu, Y.; Song, Y.; Yu, L.; Yang, B.; Mu, J.; Huang, Z.; Han, Y. Effects of different cross-linking conditions on the properties of genipin-cross-linked chitosan/collagen scaffolds for cartilage tissue engineering. J. Mater. Sci. Mater. Med. 2011, 22, 51–62. [Google Scholar]
- Khan, S.; Ranjha, N.M. Effect of degree of cross-linking on swelling and on drug release of low viscous chitosan/poly(vinyl alcohol) hydrogels. Polym. Bull. 2014, 71, 2133–2158. [Google Scholar]
- Gancheva, T.; Virgilio, N. Enhancing and Tuning the Response of Environmentally Sensitive Hydrogels with Embedded and Interconnected Pore Networks. Macromolecules 2016, 49, 5866–5876. [Google Scholar]
- Nanda, S.; Sood, N.; Reddy, B.V.K.; Markandeywa, T.S. Preparation and Characterization of Poly(vinyl alcohol)-chondroitin Sulphate Hydrogel as Scaffolds for Articular Cartilage Regeneration. Indian J. Eng. Mater. Sci. 2013, 2013, 516021. [Google Scholar]
- Mirzaei, B.E.; Ramazani, S.A.A.; Shafiee, M.; Danaei, M. Studies on Glutaraldehyde Crosslinked Chitosan Hydrogel Properties for Drug Delivery Systems. Int. J. Polym. Mater. 2013, 62, 605–611. [Google Scholar] [CrossRef]
- Kon, M.; de Visser, A.C. A poly(HEMA) sponge for restoration of articular cartilage defects. Plast. Reconstr. Surg. 1981, 67, 288–294. [Google Scholar] [CrossRef] [PubMed]
- Lund, M.N.; Luxford, C.; Skibsted, L.H.; Davies, M.J. Oxidation of myosin by haem proteins generates myosin radicals and protein cross-links. Biochem. J. 2008, 410, 565–574. [Google Scholar] [PubMed] [Green Version]
- Li, P.; Su, X.; Hao, D.; Yang, M.; Gui, T.; Cong, W.; Guo, X. One-pot method for preparation of capsaicin-containing double-network hydrogels for marine antifouling. RSC Adv. 2022, 12, 15613–15622. [Google Scholar] [PubMed]
- Ahmad, S.; Manzoor, K.; Purwar, R.; Ikram, S. Morphological and Swelling Potential Evaluation of Moringa oleifera Gum/Poly(vinyl alcohol) Hydrogels as a Superabsorbent. ACS Omega 2020, 5, 17955–17961. [Google Scholar] [PubMed]
- Weng, L.; Gouldstone, A.; Wu, Y.; Chen, W. Mechanically Strong Double Network Photocrosslinked Hydrogels from N,N-Dimethylacrylamide and Glycidyl Methacrylated Hyaluronan. Biomaterials 2008, 29, 2153–2163. [Google Scholar]
- Ronca, A.; Amora, U.D.; Raucci, M.G.; Lin, H.; Fan, Y.; Zhang, X.; Ambrosio, L. A Combined Approach of Double Network Hydrogel and Nanocomposites Based on Hyaluronic Acid and Poly(ethylene glycol) Diacrylate Blend. Materials 2018, 11, 2454. [Google Scholar] [CrossRef] [Green Version]
- Li, Z.; Wu, C.; Liu, Z.; Li, Z.; Peng, X.; Huang, J.; Ren, J.; Wang, P. A polypropylene mesh coated with interpenetrating double network hydrogel for local drug delivery in temporary closure of open abdomen. RSC Adv. 2020, 10, 1331–1340. [Google Scholar]
- Lv, B.; Bu, X.; Da, Y.; Duan, P.; Wang, H.; Ren, J.; Lyu, B.; Gao, D.; Ma, J. Gelatin/PAM double network hydrogels with super-compressibility. Polymer 2020, 210, 123021. [Google Scholar]
- Pongsavee, M. Effect of borax on immune cell proliferation and sister chromatid exchange in human chromosomes. J. Occup. Med. Toxicol. 2009, 4, 27. [Google Scholar]














{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | Crosslink-Time (h) | Wall Thickness (µm) | Pore Size (µm) | Pore Count (n */100 µm2) |
|---|---|---|---|---|
| SN30 | 2 | 0.79 ± 0.06 | 5.85 ± 0.08 | 166.75 ± 8.30 |
| SN30 | 4 | 1.54 ± 0.21 | 13.51 ± 0.28 | 106.33 ± 8.49 |
| SN30 | 6 | 2.00 ± 0.31 | 18.49 ± 0.68 | 31.33 ± 5.35 |
| SN30 | 8 | 2.81 ± 0.18 | 18.70 ± 0.23 | 28.25 ± 5.30 |
| SN30 | 10 | 3.32 ± 0.16 | 19.97 ± 0.34 | 26.25 ± 1.89 |
| SN30 | 24 | 3.77 ± 0.81 | 23.15 ± 0.47 | 20.75 ± 2.79 |
| Sample | GA Content (mL) | Wall Thickness (µm) | Pore Size (µm) | Pore Count (n/100 µm2) |
|---|---|---|---|---|
| SN10 | 10 | 1.51 ± 0.26 | 16.27 ± 0.38 | 25.33 ± 4.79 |
| SN20 | 20 | 2.07 ± 0.27 | 17.08 ± 0.22 | 23.75 ± 5.01 |
| SN30 | 30 | 3.77 ± 0.81 | 23.15 ± 0.47 | 20.75 ± 2.79 |
| SN40 | 40 | 3.91 ± 0.33 | 23.35 ± 0.93 | 18.25 ± 1.71 |
| SN50 | 50 | 5.16 ± 0.50 | 26.68 ± 0.63 | 16.50 ± 3.00 |
| Sample | Boron Content * (%) | Wall Thickness (µm) | Pore Size (µm) | Pore Count (n/100 µm2) |
|---|---|---|---|---|
| SN30 | - | 3.77 ± 0.81 | 23.15 ± 0.47 | 20.75 ± 2.79 |
| DN0.05 | 6.57 ± 0.31 | 5.86 ± 0.72 | 55.18 ± 0.77 | 2.49 ± 0.61 |
| DN0.10 | 8.73 ± 0.49 | 7.11 ± 0.66 | 53.55 ± 0.34 | 3.33 ± 0.58 |
| DN0.15 | 9.70 ± 0.50 | 8.04 ± 0.64 | 23.31 ± 0.72 | 9.00 ± 1.63 |
| Sample | Compressive Strength at Break (kPa) | Compressive Modulus (at 5–10% Strain) (kPa) | Toughness (kJ/m3) |
|---|---|---|---|
| SN30 | 443.46 ± 11.76 * | 26.49 ± 0.69 * | 69.93 ± 2.17 * |
| DN0.05 | 429.50 ± 20.84 * | 26.85 ± 1.76 * | 77.77 ± 0.62 * |
| DN0.10 | 462.83 ± 18.18 * | 28.12 ± 2.88 * | 80.03 ± 1.75 * |
| DN0.15 | 496.73 ± 20.80 | 33.99 ± 2.71 | 90.53 ± 3.38 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sringam, J.; Pankongadisak, P.; Trongsatitkul, T.; Suppakarn, N. Improving Mechanical Properties of Starch-Based Hydrogels Using Double Network Strategy. Polymers 2022, 14, 3552. https://doi.org/10.3390/polym14173552
Sringam J, Pankongadisak P, Trongsatitkul T, Suppakarn N. Improving Mechanical Properties of Starch-Based Hydrogels Using Double Network Strategy. Polymers. 2022; 14(17):3552. https://doi.org/10.3390/polym14173552
Chicago/Turabian StyleSringam, Jiradet, Porntipa Pankongadisak, Tatiya Trongsatitkul, and Nitinat Suppakarn. 2022. "Improving Mechanical Properties of Starch-Based Hydrogels Using Double Network Strategy" Polymers 14, no. 17: 3552. https://doi.org/10.3390/polym14173552