
Design, Synthesis and Characterization of Vitrimers with Low Topology Freezing Transition Temperature


Abstract
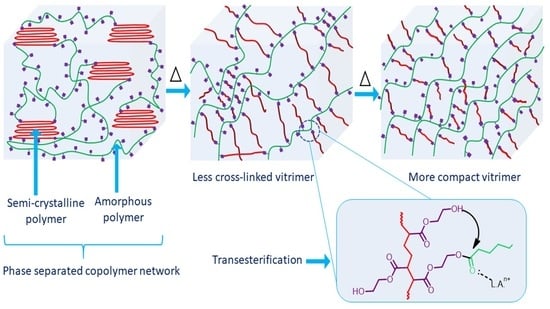
:
{kind=link}
{kind=link}
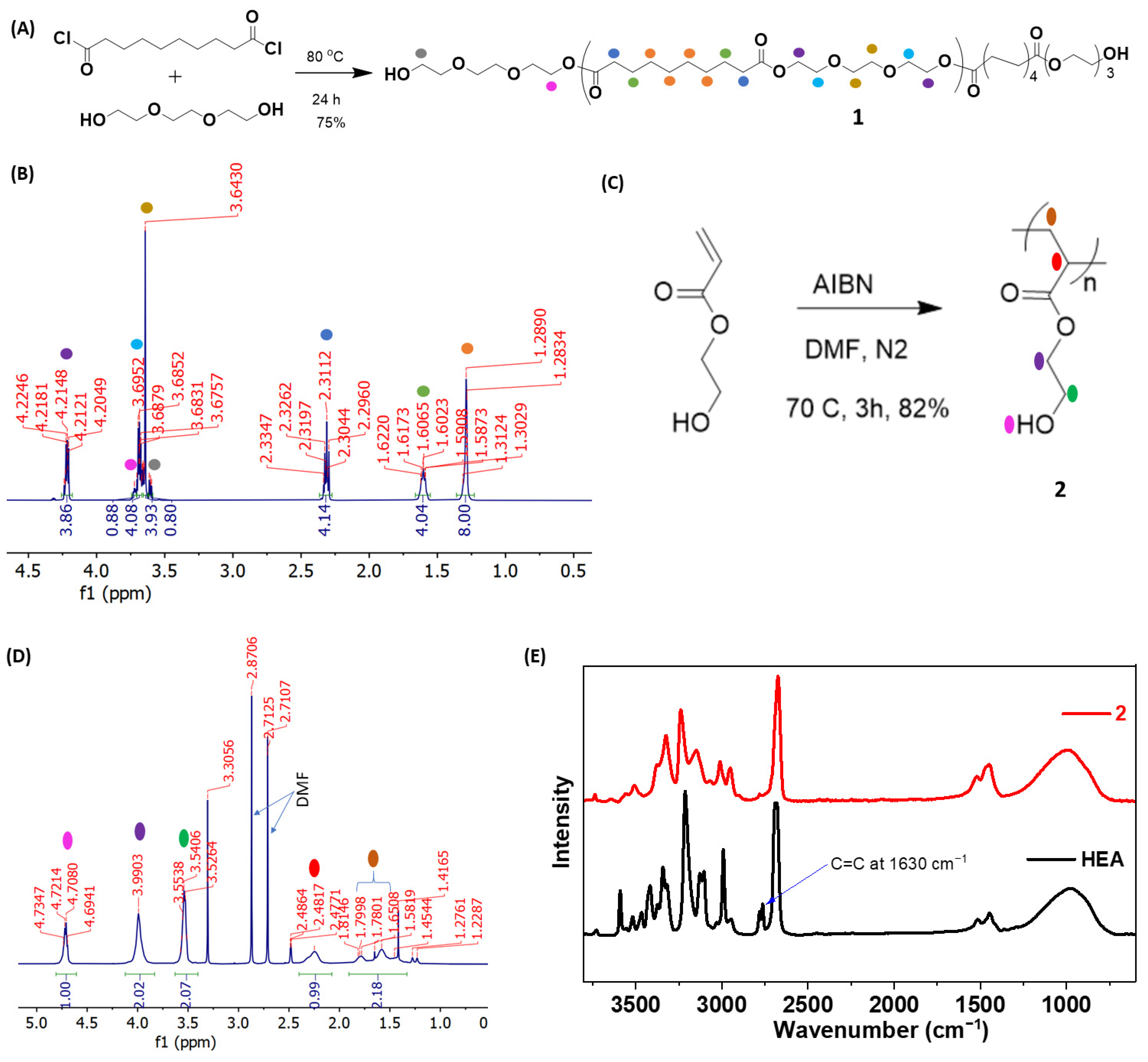{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
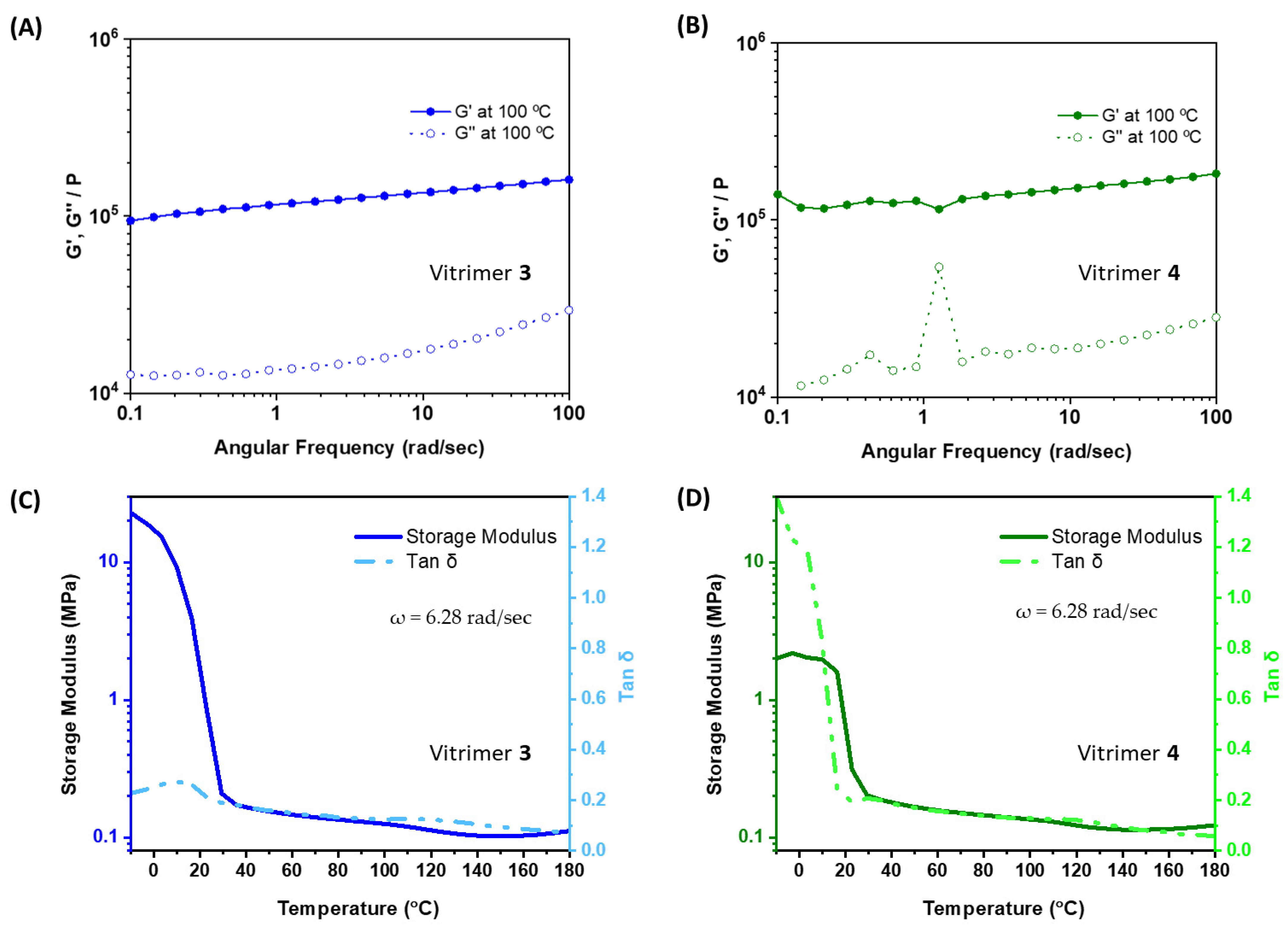{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Characterization
2.3. Synthesis of Poly(Trithylene Glycol Sebacate) (1)
2.4. Synthesis of Poly(Hydroxyethyl Acrylate) (2)
2.5. Synthesis of the Vitrimers 3 and 4
3. Results and Discussions
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Kloxin, C.J.; Bowman, C.N. Covalent adaptable networks: Smart, reconfigurable and responsive network systems. Chem. Soc. Rev. 2013, 42, 7161–7173. [Google Scholar] [CrossRef] [Green Version]
- Kloxin, C.J.; Scott, T.F.; Adzima, B.J.; Bowman, C.N. Covalent Adaptable Networks (CANs): A Unique Paradigm in Cross-Linked Polymers. Macromolecules 2010, 43, 2643–2653. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khan, A.; Ahmed, N.; Rabnawaz, M. Covalent Adaptable Network and Self-Healing Materials: Current Trends and Future Prospects in Sustainability. Polymers 2020, 12, 2027. [Google Scholar]
- Podgórski, M.; Fairbanks, B.D.; Kirkpatrick, B.E.; McBride, M.; Martinez, A.; Dobson, A.; Bongiardina, N.J.; Bowman, C.N. Toward Stimuli-Responsive Dynamic Thermosets through Continuous Development and Improvements in Covalent Adaptable Networks (CANs). Adv. Mater. 2020, 32, 1906876. [Google Scholar] [CrossRef] [PubMed]
- Winne, J.M.; Leibler, L.; Du Prez, F.E. Dynamic covalent chemistry in polymer networks: A mechanistic perspective. Polym. Chem. 2019, 10, 6091–6108. [Google Scholar] [CrossRef]
- Chen, X.; Dam, M.A.; Ono, K.; Mal, A.; Shen, H.; Nutt, S.R.; Sheran, K.; Wudl, F. A Thermally Re-mendable Cross-Linked Polymeric Material. Science 2002, 295, 1698–1702. [Google Scholar] [CrossRef]
- Green, M.S.; Tobolsky, A.V. A New Approach to the Theory of Relaxing Polymeric Media. J. Chem. Phys. 1946, 14, 80–92. [Google Scholar] [CrossRef]
- Scott, T.F.; Schneider, A.D.; Cook, W.D.; Bowman, C.N. Photoinduced Plasticity in Cross-Linked Polymers. Science 2005, 308, 1615–1617. [Google Scholar] [CrossRef]
- Kloxin, C.J.; Scott, T.F.; Park, H.Y.; Bowman, C.N. Mechanophotopatterning on a Photoresponsive Elastomer. Adv. Mater. 2011, 23, 1977–1981. [Google Scholar] [CrossRef]
- Nicolaÿ, R.; Kamada, J.; Van Wassen, A.; Matyjaszewski, K. Responsive Gels Based on a Dynamic Covalent Trithiocarbonate Cross-Linker. Macromolecules 2010, 43, 4355–4361. [Google Scholar] [CrossRef]
- Amamoto, Y.; Kamada, J.; Otsuka, H.; Takahara, A.; Matyjaszewski, K. Repeatable Photoinduced Self-Healing of Covalently Cross-Linked Polymers through Reshuffling of Trithiocarbonate Units. Angew. Chem. Int. Ed. 2011, 50, 1660–1663. [Google Scholar] [CrossRef]
- Amamoto, Y.; Otsuka, H.; Takahara, A.; Matyjaszewski, K. Self-Healing of Covalently Cross-Linked Polymers by Reshuffling Thiuram Disulfide Moieties in Air under Visible Light. Adv. Mater. 2012, 24, 3975–3980. [Google Scholar] [CrossRef]
- Montarnal, D.; Capelot, M.; Tournilhac, F.; Leibler, L. Silica-Like Malleable Materials from Permanent Organic Networks. Science 2011, 334, 965–968. [Google Scholar] [CrossRef]
- Denissen, W.; Droesbeke, M.; Nicolaÿ, R.; Leibler, L.; Winne, J.M.; Du Prez, F.E. Chemical control of the viscoelastic properties of vinylogous urethane vitrimers. Nat. Commun. 2017, 8, 14857. [Google Scholar] [CrossRef] [Green Version]
- Berne, D.; Cuminet, F.; Lemouzy, S.; Joly-Duhamel, C.; Poli, R.; Caillol, S.; Leclerc, E.; Ladmiral, V. Catalyst-Free Epoxy Vitrimers Based on Transesterification Internally Activated by an α–CF3 Group. Macromolecules 2022, 55, 1669–1679. [Google Scholar] [CrossRef]
- Bhusal, S.; Oh, C.; Kang, Y.; Varshney, V.; Ren, Y.; Nepal, D.; Roy, A.; Kedziora, G. Transesterification in Vitrimer Polymers Using Bifunctional Catalysts: Modeled with Solution-Phase Experimental Rates and Theoretical Analysis of Efficiency and Mechanisms. J. Phys. Chem. B 2021, 125, 2411–2424. [Google Scholar] [CrossRef]
- Brutman, J.P.; Delgado, P.A.; Hillmyer, M.A. Polylactide Vitrimers. ACS Macro Lett. 2014, 3, 607–610. [Google Scholar] [CrossRef] [Green Version]
- Capelot, M.; Montarnal, D.; Tournilhac, F.; Leibler, L. Metal-Catalyzed Transesterification for Healing and Assembling of Thermosets. J. Am. Chem. Soc. 2012, 134, 7664–7667. [Google Scholar] [CrossRef]
- Belowich, M.E.; Stoddart, J.F. Dynamic imine chemistry. Chem. Soc. Rev. 2012, 41, 2003–2024. [Google Scholar] [CrossRef]
- Denissen, W.; Rivero, G.; Nicolaÿ, R.; Leibler, L.; Winne, J.M.; Du Prez, F.E. Vinylogous Urethane Vitrimers. Adv. Funct. Mater. 2015, 25, 2451–2457. [Google Scholar] [CrossRef]
- Denissen, W.; De Baere, I.; Van Paepegem, W.; Leibler, L.; Winne, J.; Du Prez, F.E. Vinylogous Urea Vitrimers and Their Application in Fiber Reinforced Composites. Macromolecules 2018, 51, 2054–2064. [Google Scholar] [CrossRef] [Green Version]
- Hendriks, B.; Waelkens, J.; Winne, J.M.; Du Prez, F.E. Poly(thioether) Vitrimers via Transalkylation of Trialkylsulfonium Salts. ACS Macro Lett. 2017, 6, 930–934. [Google Scholar] [CrossRef]
- Hayashi, M.; Chen, L. Functionalization of triblock copolymer elastomers by cross-linking the end blocks via trans-N-alkylation-based exchangeable bonds. Polym. Chem. 2020, 11, 1713–1719. [Google Scholar] [CrossRef]
- Obadia, M.M.; Mudraboyina, B.P.; Serghei, A.; Montarnal, D.; Drockenmuller, E. Reprocessing and Recycling of Highly Cross-Linked Ion-Conducting Networks through Transalkylation Exchanges of C–N Bonds. J. Am. Chem. Soc. 2015, 137, 6078–6083. [Google Scholar] [CrossRef]
- Obadia, M.M.; Jourdain, A.; Cassagnau, P.; Montarnal, D.; Drockenmuller, E. Tuning the Viscosity Profile of Ionic Vitrimers Incorporating 1,2,3-Triazolium Cross-Links. Adv. Funct. Mater. 2017, 27, 1703258. [Google Scholar] [CrossRef]
- Hayashi, M.; Oba, Y.; Kimura, T.; Takasu, A. Simple preparation, properties, and functions of vitrimer-like polyacrylate elastomers using trans-N-alkylation bond exchange. Polym. J. 2021, 53, 835–840. [Google Scholar] [CrossRef]
- Denissen, W.; Winne, J.M.; Du Prez, F.E. Vitrimers: Permanent organic networks with glass-like fluidity. Chem. Sci. 2016, 7, 30–38. [Google Scholar] [CrossRef] [Green Version]
- Wu, X.; Yang, X.; Yu, R.; Zhao, X.-J.; Zhang, Y.; Huang, W. A facile access to stiff epoxy vitrimers with excellent mechanical properties via siloxane equilibration. J. Mater. Chem. A 2018, 6, 10184–10188. [Google Scholar] [CrossRef]
- Lu, Y.-X.; Guan, Z. Olefin Metathesis for Effective Polymer Healing via Dynamic Exchange of Strong Carbon–Carbon Double Bonds. J. Am. Chem. Soc. 2012, 134, 14226–14231. [Google Scholar] [CrossRef]
- Martin, R.; Rekondo, A.; Ruiz de Luzuriaga, A.; Cabañero, G.; Grande, H.J.; Odriozola, I. The processability of a poly(urea-urethane) elastomer reversibly crosslinked with aromatic disulfide bridges. J. Mater. Chem. A 2014, 2, 5710–5715. [Google Scholar] [CrossRef]
- Lafont, U.; van Zeijl, H.; van der Zwaag, S. Influence of Cross-linkers on the Cohesive and Adhesive Self-Healing Ability of Polysulfide-Based Thermosets. ACS Appl. Mater. Interfaces 2012, 4, 6280–6288. [Google Scholar] [CrossRef] [PubMed]
- Canadell, J.; Goossens, H.; Klumperman, B. Self-Healing Materials Based on Disulfide Links. Macromolecules 2011, 44, 2536–2541. [Google Scholar] [CrossRef]
- El-Zaatari, B.M.; Ishibashi, J.S.A.; Kalow, J.A. Cross-linker control of vitrimer flow. Polym. Chem. 2020, 11, 5339–5345. [Google Scholar] [CrossRef]
- Yang, Y.; Zhang, S.; Zhang, X.; Gao, L.; Wei, Y.; Ji, Y. Detecting topology freezing transition temperature of vitrimers by AIE luminogens. Nat. Commun. 2019, 10, 3165. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Capelot, M.; Unterlass, M.M.; Tournilhac, F.; Leibler, L. Catalytic Control of the Vitrimer Glass Transition. ACS Macro Lett. 2012, 1, 789–792. [Google Scholar] [CrossRef] [PubMed]
- He, C.; Christensen, P.R.; Seguin, T.J.; Dailing, E.A.; Wood, B.M.; Walde, R.K.; Persson, K.A.; Russell, T.P.; Helms, B.A. Conformational Entropy as a Means to Control the Behavior of Poly(diketoenamine) Vitrimers In and Out of Equilibrium. Angew. Chem. Int. Ed. 2020, 59, 735–739. [Google Scholar] [CrossRef] [Green Version]
- Krishnakumar, B.; Sanka, R.V.S.P.; Binder, W.H.; Parthasarthy, V.; Rana, S.; Karak, N. Vitrimers: Associative dynamic covalent adaptive networks in thermoset polymers. Chem. Eng. J. 2020, 385, 123820. [Google Scholar] [CrossRef]
- Guerre, M.; Taplan, C.; Nicolaÿ, R.; Winne, J.M.; Du Prez, F.E. Fluorinated Vitrimer Elastomers with a Dual Temperature Response. J. Am. Chem. Soc. 2018, 140, 13272–13284. [Google Scholar] [CrossRef]
- Schäler, K.; Achilles, A.; Bärenwald, R.; Hackel, C.; Saalwächter, K. Dynamics in Crystallites of Poly(ε-caprolactone) As Investigated by Solid-State NMR. Macromolecules 2013, 46, 7818–7825. [Google Scholar] [CrossRef]
- Schäler, K.; Roos, M.; Micke, P.; Golitsyn, Y.; Seidlitz, A.; Thurn-Albrecht, T.; Schneider, H.; Hempel, G.; Saalwächter, K. Basic principles of static proton low-resolution spin diffusion NMR in nanophase-separated materials with mobility contrast. Solid State Nucl. Magn. Reson. 2015, 72, 50–63. [Google Scholar] [CrossRef]
- Fernández-de-Alba, C.; Jimenez, A.M.; Abbasi, M.; Kumar, S.K.; Saalwächter, K.; Baeza, G.P. On the Immobilized Polymer Fraction in Attractive Nanocomposites: Tg Gradient versus Interfacial Layer. Macromolecules 2021, 54, 10289–10299. [Google Scholar] [CrossRef]
- Heinze, M.; Horn, C.; Pospiech, D.; Boldt, R.; Kobsch, O.; Eckstein, K.; Jehnichen, D.; Voit, B.; Baudis, S.; Liska, R.; et al. Polymer Networks for Enrichment of Calcium Ions. Polymers 2021, 13, 3506. [Google Scholar] [CrossRef] [PubMed]
- Maus, A.; Hertlein, C.; Saalwächter, K. A Robust Proton NMR Method to Investigate Hard/Soft Ratios, Crystallinity, and Component Mobility in Polymers. Macromol. Chem. Phys. 2006, 207, 1150–1158. [Google Scholar] [CrossRef]
- Saalwächter, K.; Gottlieb, M.; Liu, R.; Oppermann, W. Gelation as Studied by Proton Multiple-Quantum NMR. Macromolecules 2007, 40, 1555–1561. [Google Scholar] [CrossRef] [Green Version]
- Jakisch, L.; Garaleh, M.; Schäfer, M.; Mordvinkin, A.; Saalwächter, K.; Böhme, F. Synthesis and Structural NMR Characterization of Novel PPG/PCL Conetworks Based upon Heterocomplementary Coupling Reactions. Macromol. Chem. Phys. 2018, 219, 1700327. [Google Scholar] [CrossRef]
- Gonsalves, K.E.; Chen, X.; Cameron, J.A. Degradation of nonalternating poly(ester amides). Macromolecules 1992, 25, 3309–3312. [Google Scholar] [CrossRef]
- Greesh, N.; Sanderson, R.; Hartmann, P. Preparation of poly(styrene-b-2-hydroxyethyl acrylate) block copolymer using reverse iodine transfer polymerization. J. Appl. Polym. Sci. 2012, 126, 1773–1783. [Google Scholar] [CrossRef]
- Debnath, S.; Kaushal, S.; Ojha, U. Catalyst-Free Partially Bio-Based Polyester Vitrimers. ACS Appl. Polym. Mater. 2020, 2, 1006–1013. [Google Scholar] [CrossRef]
- Foli, G.; Degli Esposti, M.; Toselli, M.; Morselli, D.; Fabbri, P. Facile method based on 19F-NMR for the determination of hydroxyl value and molecular weight of hydroxyl terminated polymers. Analyst 2019, 144, 2087–2096. [Google Scholar] [CrossRef]
- Coca, S.; Jasieczek, C.B.; Beers, K.L.; Matyjaszewski, K. Polymerization of acrylates by atom transfer radical polymerization. Homopolymerization of 2-hydroxyethyl acrylate. J. Polym. Sci. Part A Polym. Chem. 1998, 36, 1417–1424. [Google Scholar] [CrossRef]
- Takeshita, H.; Shiomi, T.; Takenaka, K.; Arai, F. Crystallization and higher-order structure of multicomponent polymeric systems. Polymer 2013, 54, 4776–4789. [Google Scholar] [CrossRef] [Green Version]
- Morita, S. Hydrogen-bonds structure in poly(2-hydroxyethyl methacrylate) studied by temperature-dependent infrared spectroscopy. Front. Chem. 2014, 2, 10. [Google Scholar] [CrossRef] [PubMed] [Green Version]










Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Krishnan, B.P.; Saalwaechter, K.; Adjedje, V.K.B.; Binder, W.H. Design, Synthesis and Characterization of Vitrimers with Low Topology Freezing Transition Temperature. Polymers 2022, 14, 2456. https://doi.org/10.3390/polym14122456
Krishnan BP, Saalwaechter K, Adjedje VKB, Binder WH. Design, Synthesis and Characterization of Vitrimers with Low Topology Freezing Transition Temperature. Polymers. 2022; 14(12):2456. https://doi.org/10.3390/polym14122456
Chicago/Turabian StyleKrishnan, Baiju P., Kay Saalwaechter, Vico K. B. Adjedje, and Wolfgang H. Binder. 2022. "Design, Synthesis and Characterization of Vitrimers with Low Topology Freezing Transition Temperature" Polymers 14, no. 12: 2456. https://doi.org/10.3390/polym14122456