
Solute Diffusivity and Local Free Volume in Cross-Linked Polymer Network: Implication of Optimizing the Conductivity of Polymer Electrolyte


Abstract
:1. Introduction
2. Methods
2.1. Coarse-Grained Molecular Simulations for Cross-Linked Polymer
2.2. Free Volume Theory for Diffusion
3. Results and Discussions
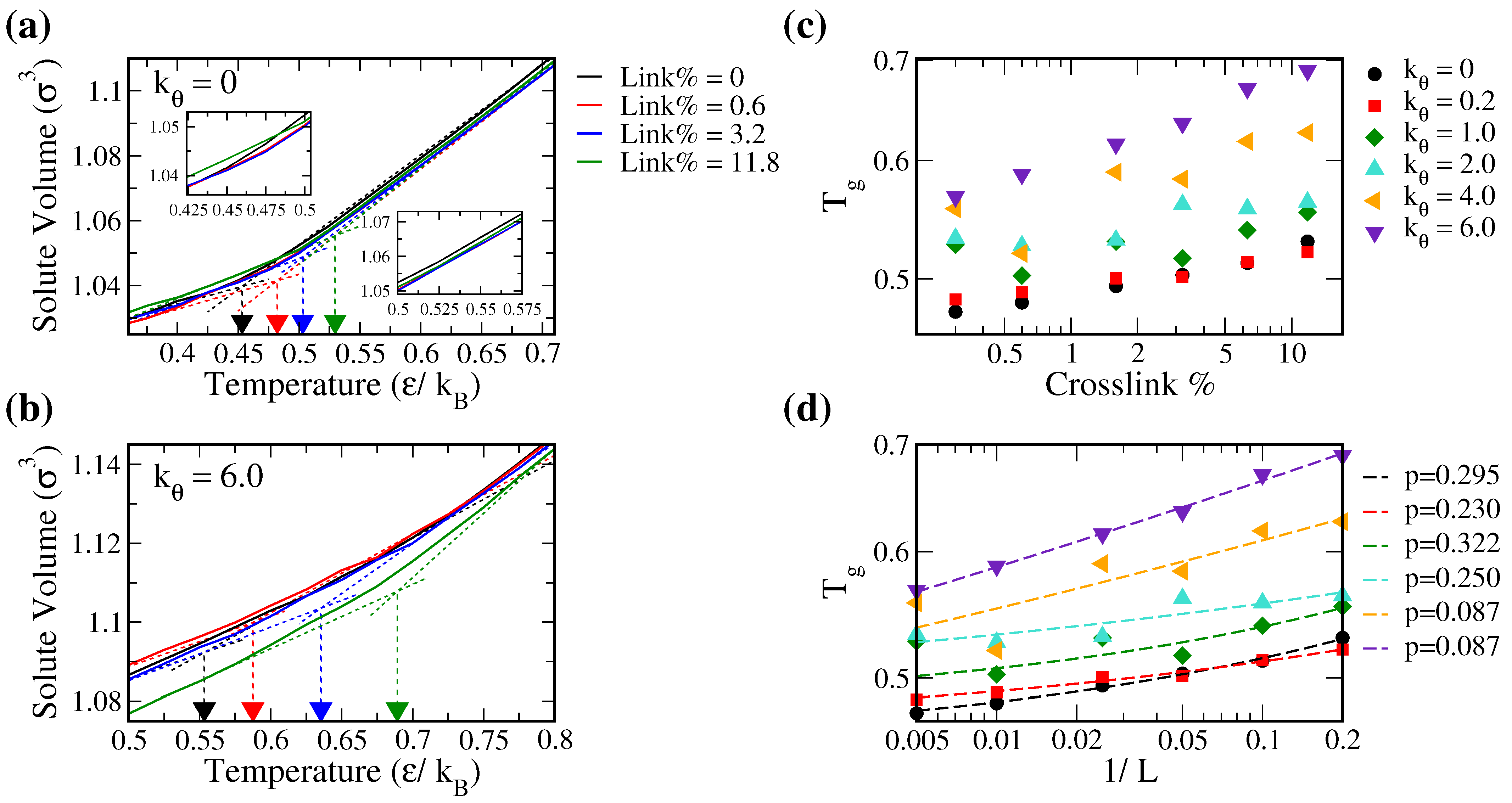
3.1. Solute Diffusion in Cross-Linked Polymers
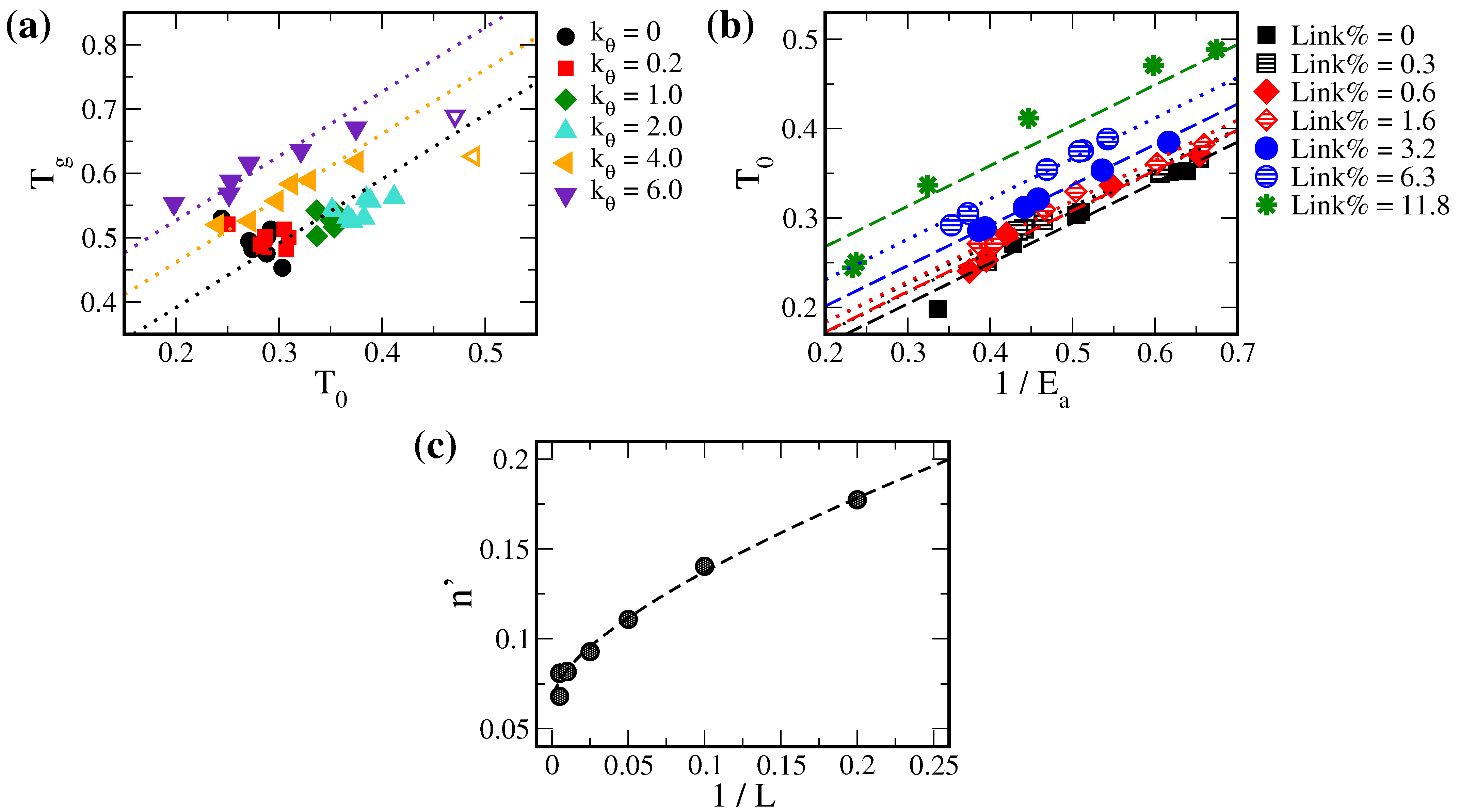
3.2. Beyond Compensation Effect in the VTF Model
3.3. Solute Diffusion and Free Volume
3.4. Optimization of Solute Diffusion in Cross-Linked Polymers
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- George, S.C.; Thomas, S. Transport phenomena through polymeric systems. Prog. Polym. Sci. 2001, 26, 985–1017. [Google Scholar] [CrossRef]
- Vesely, D. Diffusion of liquids in polymers. Int. Mater. Rev. 2013, 53, 299–315. [Google Scholar] [CrossRef]
- Sharma, J.; Tewari, K.; Arya, R.K. Diffusion in polymeric systems—A review on free volume theory. Prog. Org. Coat. 2017, 111, 83–92. [Google Scholar] [CrossRef]
- Saal, A.; Hagemann, T.; Schubert, U.S. Polymers for Battery Applications—Active Materials, Membranes, and Binders. Adv. Energy Mater. 2020, 121, 2001984. [Google Scholar] [CrossRef]
- Mindemark, J.; Lacey, M.J.; Bowden, T.; Brandell, D. Beyond PEO—Alternative host materials for Li+-conducting solid polymer electrolytes. Prog. Polym. Sci. 2018, 81, 114–143. [Google Scholar] [CrossRef]
- Mogurampelly, S.; Borodin, O.; Ganesan, V. Computer Simulations of Ion Transport in Polymer Electrolyte Membranes. Annu. Rev. Chem. Biomol. Eng. 2016, 7, 349–371. [Google Scholar] [CrossRef]
- Aziz, S.B.; Woo, T.J.; Kadir, M.; Ahmed, H.M. A conceptual review on polymer electrolytes and ion transport models. J. Sci. Adv. Mater. Devices 2018, 3, 1–17. [Google Scholar] [CrossRef]
- Hasan, N.; Pulst, M.; Samiullah, M.H.; Kressler, J. Comparison of Li+-ion conductivity in linear and crosslinked poly(ethylene oxide). J. Polym. Sci. Part B Polym. Phys. 2018, 57, 21–28. [Google Scholar] [CrossRef] [Green Version]
- Baroncini, E.A.; Rousseau, D.M.; IV, C.A.S.; Stanzione, J.F., III. Optimizing conductivity and cationic transport in crosslinked solid polymer electrolytes. Solid State Ion. 2020, 345, 115161. [Google Scholar] [CrossRef]
- Othman, L.; Chew, K.W.; Osman, Z. Impedance spectroscopy studies of poly (methyl methacrylate)-lithium salts polymer electrolyte systems. Ionics 2007, 13, 337–342. [Google Scholar] [CrossRef]
- Krongauz, V.V. Compensation effect: Sublimation, diffusion in polymers, polymer degradation. J. Therm. Anal. Calorim. 2019, 138, 3425–3444. [Google Scholar] [CrossRef]
- Zheng, J.M.; Qiu, J.; Madeira, L.M.; Mendes, A. Polymer Structure and the Compensation Effect of the Diffusion Pre-Exponential Factor and Activation Energy of a Permeating Solute. J. Phys. Chem. B 2007, 111, 2828–2835. [Google Scholar] [CrossRef] [PubMed]
- Baskaran, R.; Selvasekarapandian, S.; Kuwata, N.; Kawamura, J.; Hattori, T. ac impedance, DSC and FT-IR investigations on (x)PVAc-(1-x)PVdF blends with LiClO4. Mater. Chem. Phys. 2006, 98, 55–61. [Google Scholar] [CrossRef]
- Fulcher, G.S. Analyis of Recent Measurements of the Viscosity of Glasses. J. Am. Ceram. Soc. 1925, 8, 339–355. [Google Scholar] [CrossRef]
- MacCallum, J.R.; Vincent, C.A. Polymer Electrolyte Reviews; Elsevier Applied Science: London, UK, 1989. [Google Scholar]
- Bamford, D.; Reiche, A.; Dlubek, G.; Alloin, F.; Sanchez, J.Y.; Alam, M.A. Ionic conductivity, glass transition, and local free volume in poly(ethylene oxide) electrolytes: Single and mixed ion conductors. J. Chem. Phys. 2003, 118, 9420–9432. [Google Scholar] [CrossRef]
- Cohen, M.H.; Turnbull, D. Molecular Transport in Liquids and Glasses. J. Chem. Phys. 1959, 31, 1164–1169. [Google Scholar] [CrossRef] [Green Version]
- Turnbull, D.; Cohen, M.H. Free-Volume Model of the Amorphous Phase: Glass Transition. J. Chem. Phys. 1961, 34, 120–125. [Google Scholar] [CrossRef]
- Diederichsen, K.M.; Buss, H.G.; McCloskey, B.D. The Compensation Effect in the Vogel–Tammann–Fulcher (VTF) Equation for Polymer-Based Electrolytes. Macromolecules 2017, 50, 3831–3840. [Google Scholar] [CrossRef]
- Cowie, J. Effect of side chain length and crosslinking on the ac conductivity of oligo (ethyleneoxide) comb-branch polymer-salt mixtures. Solid State Ion. 1990, 42, 243–249. [Google Scholar] [CrossRef]
- Pan, Q.; Smith, D.M.; Qi, H.; Wang, S.; Li, C.Y. Hybrid Electrolytes with Controlled Network Structures for Lithium Metal Batteries. Adv. Mater. 2015, 27, 5995–6001. [Google Scholar] [CrossRef]
- Krongauz, V.V. Diffusion in polymers dependence on crosslink density. J. Therm. Anal. Calorim. 2010, 102, 435–445. [Google Scholar] [CrossRef]
- Zhang, J.; Li, X.; Li, Y.; Wang, H.; Ma, C.; Wang, Y.; Hu, S.; Wei, W. Cross-Linked Nanohybrid Polymer Electrolytes with POSS Cross-Linker for Solid-State Lithium Ion Batteries. Front. Chem. 2018, 6, 186. [Google Scholar] [CrossRef] [PubMed]
- Kremer, K.; Grest, G.S. Dynamics of entangled linear polymer melts: A molecular-dynamics simulation. J. Chem. Phys. 1990, 92, 5057–5086. [Google Scholar] [CrossRef]
- Nowak, C.; Escobedo, F.A. Tuning the Sawtooth Tensile Response and Toughness of Multiblock Copolymer Diamond Networks. Macromolecules 2016, 49, 6711–6721. [Google Scholar] [CrossRef]
- Aguilera-Mercado, B.M.; Cohen, C.; Escobedo, F.A. Sawtooth Tensile Response of Model Semiflexible and Block Copolymer Elastomers. Macromolecules 2014, 47, 840–850. [Google Scholar] [CrossRef]
- Kalra, V.; Escobedo, F.; Joo, Y.L. Effect of shear on nanoparticle dispersion in polymer melts: A coarse-grained molecular dynamics study. J. Chem. Phys. 2010, 132, 024901. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Cao, D.; Zhang, L. Molecular Dynamics Study on Nanoparticle Diffusion in Polymer Melts: A Test of the Stokes-Einstein Law. J. Phys. Chem. C 2008, 112, 6653–6661. [Google Scholar] [CrossRef]
- Sen, S.; Kumar, S.K.; Keblinski, P. Viscoelastic Properties of Polymer Melts from Equilibrium Molecular Dynamics Simulations. Macromolecules 2005, 38, 650–653. [Google Scholar] [CrossRef] [Green Version]
- Alshammasi, M.S.; Escobedo, F.A. Correlation between Ionic Mobility and Microstructure in Block Copolymers. A Coarse-Grained Modeling Study. Macromolecules 2018, 51, 9213–9221. [Google Scholar] [CrossRef]
- Bishop, M.; Kalos, M.H.; Frisch, H.L. Molecular dynamics of polymeric systems. J. Chem. Phys. 1979, 70, 1299–1304. [Google Scholar] [CrossRef]
- Weeks, J.D.; Chandler, D.; Andersen, H.C. Role of Repulsive Forces in Determining the Equilibrium Structure of Simple Liquids. J. Chem. Phys. 1971, 54, 5237–5247. [Google Scholar] [CrossRef]
- Xie, H.; Basu, S.; DeMeter, E.C. Molecular Dynamics Simulations of Photo-Induced Free Radical Polymerization. J. Chem. Inf. Model. 2020, 60, 6314–6327. [Google Scholar] [CrossRef] [PubMed]
- Plimpton, S. Fast Parallel Algorithms for Short-Range Molecular Dynamics. J. Comput. Phys. 1995, 117, 1–19. [Google Scholar] [CrossRef] [Green Version]
- Nosé, S. A molecular dynamics method for simulations in the canonical ensemble. Mol. Phys. 2006, 52, 255–268. [Google Scholar] [CrossRef]
- Hoover, W.G. Canonical dynamics: Equilibrium phase-space distributions. Phys. Rev. A 1985, 31, 1695–1697. [Google Scholar] [CrossRef] [Green Version]
- Nosé, S. A unified formulation of the constant temperature molecular dynamics methods. J. Chem. Phys. 1984, 81, 511–519. [Google Scholar] [CrossRef] [Green Version]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual molecular dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef]
- White, R.P.; Lipson, J.E.G. Polymer Free Volume and Its Connection to the Glass Transition. Macromolecules 2016, 49, 3987–4007. [Google Scholar] [CrossRef]
- Rycroft, C.H.; Grest, G.S.; Landry, J.W.; Bazant, M.Z. Analysis of granular flow in a pebble-bed nuclear reactor. Phys. Rev. E 2006, 74, 021306. [Google Scholar] [CrossRef] [Green Version]
- Fox, T.G.; Flory, P.J. Second-Order Transition Temperatures and Related Properties of Polystyrene. I. Influence of Molecular Weight. J. Appl. Phys. 1950, 21, 581–591. [Google Scholar] [CrossRef]
- Fox, T.G.; Flory, P.J. Further Studies on the Melt Viscosity of Polyisobutylene. J. Phys. Chem. 1951, 55, 221–234. [Google Scholar] [CrossRef] [PubMed]
- Fox, T.G.; Flory, P.J. The glass temperature and related properties of polystyrene. Influence of molecular weight. J. Polym. Sci. 1954, 14, 315–319. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Type i-j | Potential Form | ||
|---|---|---|---|
| P-P | 1.0 | 1.0 | LJ |
| C-C | 1.0 | 1.0 | WCA |
| A-A | 1.0 | 1.0 | WCA |
| P-C | 2.0 | 1.0 | LJ |
| P-A | 1.0 | 1.0 | LJ |
| C-A | 2.0 | 1.0 | LJ |
| Link% | Polymer Chain Length (L) × Chain Number | Reactive Polymer Chain Length (L+2) × Chain Number | ||||
|---|---|---|---|---|---|---|
| 0 | 30,000 | 0 | 1875 | 1875 | ||
| 0.3 | 30,000 | 100 | 1875 | 1875 | ||
| 0.6 | 30,000 | 200 | 1875 | 1875 | ||
| 1.6 | 30,000 | 500 | 1875 | 1875 | ||
| 3.2 | 30,000 | 1000 | 1875 | 1875 | ||
| 6.3 | 30,000 | 2000 | 1875 | 1875 | ||
| 11.8 | 30,000 | 4000 | 1875 | 1875 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tsai, Y.-C.; Chiu, C.-C. Solute Diffusivity and Local Free Volume in Cross-Linked Polymer Network: Implication of Optimizing the Conductivity of Polymer Electrolyte. Polymers 2022, 14, 2061. https://doi.org/10.3390/polym14102061
Tsai Y-C, Chiu C-C. Solute Diffusivity and Local Free Volume in Cross-Linked Polymer Network: Implication of Optimizing the Conductivity of Polymer Electrolyte. Polymers. 2022; 14(10):2061. https://doi.org/10.3390/polym14102061
Chicago/Turabian StyleTsai, Yi-Chen, and Chi-Cheng Chiu. 2022. "Solute Diffusivity and Local Free Volume in Cross-Linked Polymer Network: Implication of Optimizing the Conductivity of Polymer Electrolyte" Polymers 14, no. 10: 2061. https://doi.org/10.3390/polym14102061