
Interfacial Phenomena in Multi-Micro-/Nanolayered Polymer Coextrusion: A Review of Fundamental and Engineering Aspects



Abstract
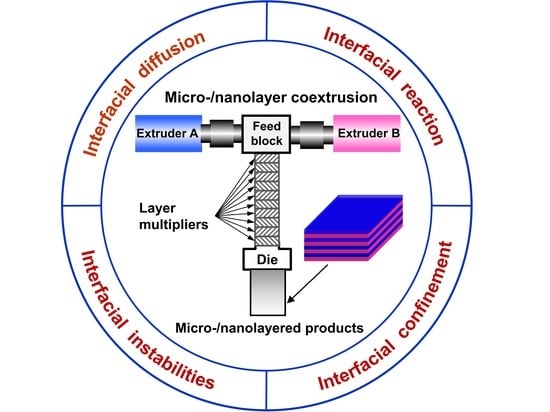
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Interfacial Phenomena at Polymer–Polymer Interfaces
2.1. Interfacial Diffusion
2.1.1. Basic Interdiffusion Theories
2.1.2. Interdiffusion in Micro-/Nanolayer Coextrusion
2.2. Interfacial Instabilities
2.2.1. Interfacial Flow Instabilities
2.2.2. Interfacial Slip
2.2.3. Layer Breakup in Nanolayer Coextrusion
2.3. Interfacial Reaction
2.3.1. Basic Theories
2.3.2. Interfacial Morphology under Reaction
Reaction under Equilibrium Conditions
Morphology Development Revealed by Rheometry
Effects of Oscillatory Shear Flow
2.3.3. Interfacial Reaction in Multilayer Coextrusion
2.4. Interfacial Confinement in Micro-/Nanolayer Coextrusion
2.4.1. Confined Crystallization
2.4.2. Confined Glass Transition Dynamics
3. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Qiu, H.; Bousmina, M. New technique allowing the quantification of diffusion at polymer/polymer interfaces using rheological analysis: Theoretical and experimental results. J. Rheol. 1999, 43, 551–568. [Google Scholar] [CrossRef]
- Macosko, C.W.; Jeon, H.K.; Hoye, T.R. Reactions at polymer–polymer interfaces for blend compatibilization. Prog. Polym. Sci. 2005, 30, 939–947. [Google Scholar] [CrossRef]
- Zhao, R.; Macosko, C.W. Slip at polymer–polymer interfaces: Rheological measurements on coextruded multilayers. J. Rheol. 2002, 46, 145–167. [Google Scholar] [CrossRef]
- Lai, C.-Y.; Ponting, M.T.; Baer, E. Influence of interdiffusion on multilayered gradient refractive index (GRIN) lens materials. Polymer 2012, 53, 1393–1403. [Google Scholar] [CrossRef]
- Lu, B.; Lamnawar, K.; Maazouz, A.; Sudre, G. Critical Role of Interfacial Diffusion and Diffuse Interphases Formed in Multi-Micro-/Nanolayered Polymer Films Based on Poly(vinylidene fluoride) and Poly(methyl methacrylate). ACS Appl. Mater. Interfaces 2018, 10, 29019–29037. [Google Scholar] [CrossRef] [Green Version]
- Sun, Y.-J.; Hu, G.-H.; Lambla, M.; Kotlar, H.K. In situ compatibilization of polypropylene and poly(butylene terephthalate) polymer blends by one-step reactive extrusion. Polymer 1996, 37, 4119–4127. [Google Scholar] [CrossRef]
- Jiang, G.; Wu, H.; Guo, S. Reinforcement of adhesion and development of morphology at polymer–polymer interface via reactive compatibilization: A review. Polym. Eng. Sci. 2010, 50, 2273–2286. [Google Scholar] [CrossRef]
- Richardson, J.J.; Cui, J.; Björnmalm, M.; Braunger, J.A.; Ejima, H.; Caruso, F. Innovation in Layer-by-Layer Assembly. Chem. Rev. 2016, 116, 14828–14867. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schrenk, W. Method for Multilayer Coextrusion. U.S. Patent 3,773,882 A, 20 November 1973. [Google Scholar]
- Dooley, J. Viscoelastic Flow Effects in Multilayer Polymer Coextrusion. Ph.D. Thesis, Eindhoven University of Technology, Eindhoven, The Netherlands, 2002. [Google Scholar]
- Han, C.D. Multiphase Flow in Polymer Processing. In Rheology: Volume 3: Applications; Astarita, G., Marrucci, G., Nicolais, L., Eds.; Springer US: Boston, MA, USA, 1980; pp. 121–128. [Google Scholar]
- Langhe, D.; Ponting, M. Manufacturing and Novel Applications of Multilayer Polymer Films; Elsevier: Oxford, UK, 2016. [Google Scholar]
- Lu, B.; Alcouffe, P.; Sudre, G.; Pruvost, S.; Serghei, A.; Liu, C.; Maazouz, A.; Lamnawar, K. Unveiling the Effects of In Situ Layer–Layer Interfacial Reaction in Multilayer Polymer Films via Multilayered Assembly: From Microlayers to Nanolayers. Macromol. Mater. Eng. 2020, 305, 2000076. [Google Scholar] [CrossRef]
- Mueller, C.D.; Nazarenko, S.; Ebeling, T.; Schuman, T.L.; Hiltner, A.; Baer, E. Novel structures by microlayer coextrusion—talc-filled PP, PC/SAN, and HDPE/LLDPE. Polym. Eng. Sci. 1997, 37, 355–362. [Google Scholar] [CrossRef]
- Mitsoulis, E.; Wagner, R.; Heng, F.L. Numerical simulation of wire-coating low-density polyethylene: Theory and experiments. Polym. Eng. Sci. 1988, 28, 291–310. [Google Scholar] [CrossRef]
- Mitsoulis, E. Multilayer Film Coextrusion of Polymer Melts: Analysis of Industrial Lines with the Finite Element Method. J. Polym. Eng. 2005, 25, 393–410. [Google Scholar] [CrossRef]
- Valette, R.; Laure, P.; Demay, Y.; Agassant, J.F. Convective linear stability analysis of two-layer coextrusion flow for molten polymers. J. Non-Newton. Fluid Mech. 2004, 121, 41–53. [Google Scholar] [CrossRef]
- Lamnawar, K.; Maazouz, A. Role of the interphase in the flow stability of reactive coextruded multilayer polymers. Polym. Eng. Sci. 2009, 49, 727–739. [Google Scholar] [CrossRef]
- Wilson, G.M.; Khomami, B. An experimental investigation of interfacial instabilities in multilayer flow of viscoelastic fluids. III. Compatible polymer systems. J. Rheol. 1993, 37, 341–354. [Google Scholar] [CrossRef]
- Ponting, M.; Hiltner, A.; Baer, E. Polymer Nanostructures by Forced Assembly: Process, Structure, and Properties. Macromol. Symp. 2010, 294, 19–32. [Google Scholar] [CrossRef]
- Burt, T.M.; Jordan, A.M.; Korley, L.T.J. Toward Anisotropic Materials via Forced Assembly Coextrusion. ACS Appl. Mater. Interfaces 2012, 4, 5155–5161. [Google Scholar] [CrossRef]
- Burt, T.M.; Keum, J.; Hiltner, A.; Baer, E.; Korley, L.T.J. Confinement of Elastomeric Block Copolymers via Forced Assembly Coextrusion. ACS Appl. Mater. Interfaces 2011, 3, 4804–4811. [Google Scholar] [CrossRef]
- Lai, C.-Y.; Hiltner, A.; Baer, E.; Korley, L.T.J. Deformation of Confined Poly(ethylene oxide) in Multilayer Films. ACS Appl. Mater. Interfaces 2012, 4, 2218–2227. [Google Scholar] [CrossRef]
- Baer, E.; Zhu, L. 50th Anniversary Perspective: Dielectric Phenomena in Polymers and Multilayered Dielectric Films. Macromolecules 2017, 50, 2239–2256. [Google Scholar] [CrossRef]
- Carr, J.M.; Langhe, D.S.; Ponting, M.T.; Hiltner, A.; Baer, E. Confined Crystallization in Polymer Nanolayered Films: A review. J. Mater. Res. 2012, 27, 1326–1350. [Google Scholar] [CrossRef] [Green Version]
- Aradian, A.; Raphaël, E.; de Gennes, P.G. Strengthening of a Polymer Interface: Interdiffusion and Cross-Linking. Macromolecules 2000, 33, 9444–9451. [Google Scholar] [CrossRef] [Green Version]
- Du, W.; Yuan, G.; Wang, M.; Han, C.C.; Satija, S.K.; Akgun, B. Initial Stages of Interdiffusion between Asymmetrical Polymeric Layers: Glassy Polycarbonate and Melt Poly(methyl methacrylate) Interface Studied by Neutron Reflectometry. Macromolecules 2014, 47, 713–720. [Google Scholar] [CrossRef]
- Kim, J.K.; Han, C.D. Polymer-polymer interdiffusion during coextrusion. Polym. Eng. Sci. 1991, 31, 258–269. [Google Scholar] [CrossRef]
- Yang, L.; Suo, T.; Niu, Y.; Wang, Z.; Yan, D.; Wang, H. Effects of phase behavior on mutual diffusion at polymer layers interface. Polymer 2010, 51, 5276–5281. [Google Scholar] [CrossRef]
- Shi, M.; Zhang, Y.; Cheng, L.; Jiao, Z.; Yang, W.; Tan, J.; Ding, Y. Interfacial Diffusion and Bonding in Multilayer Polymer Films: A Molecular Dynamics Simulation. J. Phys. Chem. B 2016, 120, 10018–10029. [Google Scholar] [CrossRef]
- Zhao, R.; Macosko, C.W. Polymer–polymer mutual diffusion via rheology of coextruded multilayers. AlChE J. 2007, 53, 978–985. [Google Scholar] [CrossRef]
- Liu, R.Y.F.; Jin, Y.; Hiltner, A.; Baer, E. Probing Nanoscale Polymer Interactions by Forced-Assembly. Macromol. Rapid Commun. 2003, 24, 943–948. [Google Scholar] [CrossRef]
- Liu, R.Y.F.; Ranade, A.P.; Wang, H.P.; Bernal-Lara, T.E.; Hiltner, A.; Baer, E. Forced Assembly of Polymer Nanolayers Thinner Than the Interphase. Macromolecules 2005, 38, 10721–10727. [Google Scholar] [CrossRef]
- Ji, X.; Chen, D.; Zheng, Y.; Shen, J.; Guo, S.; Harkin-Jones, E. Multilayered assembly of poly(vinylidene fluoride) and poly(methyl methacrylate) for achieving multi-shape memory effects. Chem. Eng. J. 2019, 362, 190–198. [Google Scholar] [CrossRef]
- Decker, J.J.; Meyers, K.P.; Paul, D.R.; Schiraldi, D.A.; Hiltner, A.; Nazarenko, S. Polyethylene-based nanocomposites containing organoclay: A new approach to enhance gas barrier via multilayer coextrusion and interdiffusion. Polymer 2015, 61, 42–54. [Google Scholar] [CrossRef]
- Yin, K.; Zhou, Z.; Schuele, D.E.; Wolak, M.; Zhu, L.; Baer, E. Effects of Interphase Modification and Biaxial Orientation on Dielectric Properties of Poly(ethylene terephthalate)/Poly(vinylidene fluoride-co-hexafluoropropylene) Multilayer Films. ACS Appl. Mater. Interfaces 2016, 8, 13555–13566. [Google Scholar] [CrossRef] [PubMed]
- Composto, R.J.; Kramer, E.J.; White, D.M. Mutual diffusion in the miscible polymer blend polystyrene/poly(xylenyl ether). Macromolecules 1988, 21, 2580–2588. [Google Scholar] [CrossRef]
- Kramer, E.J.; Green, P.; Palmstrøm, C.J. Interdiffusion and Marker Movements in Concentrated Polymer-Polymer Diffusion Couples. Polymer 1984, 25, 473–480. [Google Scholar] [CrossRef]
- Brochard, F.; Jouffroy, J.; Levinson, P. Polymer-polymer diffusion in melts. Macromolecules 1983, 16, 1638–1641. [Google Scholar] [CrossRef]
- Langhe, D.; Ponting, M. Gas Transport, Mechanical, Interphase, and Interdiffusion Properties in Coextruded-Multilayered Films. In Manufacturing and Novel Applications of Multilayer Polymer Films; Langhe, D., Ponting, M., Eds.; William Andrew Publishing: Boston, MA, USA, 2016; pp. 46–116. [Google Scholar]
- Pollock, G.; Nazarenko, S.; Hiltner, A.; Baer, E. Interdiffusion in Microlayered Polymer Composites of Polycarbonate and a Copolyester. J. Appl. Polym. Sci. 1994, 52, 163–176. [Google Scholar] [CrossRef]
- Qiu, H.; Bousmina, M. Determination of Mutual Diffusion Coefficients at Nonsymmetric Polymer/Polymer Interfaces from Rheometry. Macromolecules 2000, 33, 6588–6594. [Google Scholar] [CrossRef]
- Zhang, H.; Lamnawar, K.; Maazouz, A. Rheological Modeling of the Mutual Diffusion and the Interphase Development for an Asymmetrical Bilayer Based on PMMA and PVDF Model Compatible Polymers. Macromolecules 2013, 46, 276–299. [Google Scholar] [CrossRef]
- Zhang, H.; Lamnawar, K.; Maazouz, A. Fundamental Understanding and Modeling of Diffuse Interphase Properties and Its Role in Interfacial Flow Stability of Multilayer Polymers. Polym. Eng. Sci. 2015, 55, 771–791. [Google Scholar] [CrossRef]
- Zhang, H.; Lamnawar, K.; Maazouz, A.; Maia, J.M. A Nonlinear Shear and Elongation Rheological Study of Interfacial Failure in Compatible Bilayer Systems. J. Rheol. 2016, 60, 1–23. [Google Scholar] [CrossRef]
- Zhang, H.; Lamnawar, K.; Maazouz, A. Understanding of Transient Rheology in Step Shear and Its Implication to Explore Nonlinear Relaxation Dynamics of Interphase in Compatible Polymer Multi-microlayered Systems. Ind. Eng. Chem. Res. 2018, 57, 8093–8104. [Google Scholar] [CrossRef]
- Arendt, B.H.; Krishnamoorti, R.; Kornfield, J.A.; Smith, S.D. Component Dynamics in Miscible Blends: Equally and Unequally Entangled Polyisoprene/Polyvinylethylene. Macromolecules 1997, 30, 1127–1137. [Google Scholar] [CrossRef]
- Schrenk, W.J.; Bradley, N.L.; Alfrey, T.; Maack, H. Interfacial flow instability in multilayer coextrusion. Polym. Eng. Sci. 1978, 18, 620–623. [Google Scholar] [CrossRef]
- Han, C.D.; Shetty, R. Studies on multilayer film coextrusion II. Interfacial instability in flat film coextrusion. Polym. Eng. Sci. 1978, 18, 180–186. [Google Scholar] [CrossRef]
- Ramanathan, R.; Shanker, R.; Rehg, T.; Jons, S.; Headley, D.; Schrenk, W. “Wave” Pattern Instability in Multilayer Coextrusion-An Experimental Investigation; SPE ANTEC Proc; Society of Plastics Engineers Inc.: Indianapolis, IN, USA, 1996; pp. 224–228. [Google Scholar]
- Bironeau, A.; Salez, T.; Miquelard-Garnier, G.; Sollogoub, C. Existence of a Critical Layer Thickness in PS/PMMA Nanolayered Films. Macromolecules 2017, 50, 4064–4073. [Google Scholar] [CrossRef] [Green Version]
- Jordan, A.M.; Lee, B.; Kim, K.; Ludtke, E.; Lhost, O.; Jaffer, S.A.; Bates, F.S.; Macosko, C.W. Rheology of polymer multilayers: Slip in shear, hardening in extension. J. Rheol. 2019, 63, 751–761. [Google Scholar] [CrossRef]
- Lee, P.C.; Park, H.E.; Morse, D.C.; Macosko, C.W. Polymer-polymer interfacial slip in multilayered films. J. Rheol. 2009, 53, 893–915. [Google Scholar] [CrossRef]
- Lee, P.C.; Macosko, C.W. Polymer-polymer interfacial slip by direct visualization and by stress reduction. J. Rheol. 2010, 54, 1207–1218. [Google Scholar] [CrossRef]
- Bernal-Lara, T.E.; Liu, R.Y.F.; Hiltner, A.; Baer, E. Structure and thermal stability of polyethylene nanolayers. Polymer 2005, 46, 3043–3055. [Google Scholar] [CrossRef]
- Scholtyssek, S.; Adhikari, R.; Seydewitz, V.; Michler, G.H.; Baer, E.; Hiltner, A. Evaluation of Morphology and Deformation Micromechanisms in Multilayered PP/PS Films: An Electron Microscopy Study. Macromol. Symp. 2010, 294, 33–44. [Google Scholar] [CrossRef]
- Zhu, Y.; Bironeau, A.; Restagno, F.; Sollogoub, C.; Miquelard-Garnier, G. Kinetics of thin polymer film rupture: Model experiments for a better understanding of layer breakups in the multilayer coextrusion process. Polymer 2016, 90 (Suppl. C), 156–164. [Google Scholar] [CrossRef] [Green Version]
- Lin, Y.; Hiltner, A.; Baer, E. A new method for achieving nanoscale reinforcement of biaxially oriented polypropylene film. Polymer 2010, 51, 4218–4224. [Google Scholar] [CrossRef]
- Feng, J.; Zhang, Z.; Bironeau, A.; Guinault, A.; Miquelard-Garnier, G.; Sollogoub, C.; Olah, A.; Baer, E. Breakup behavior of nanolayers in polymeric multilayer systems—Creation of nanosheets and nanodroplets. Polymer 2018, 143, 19–27. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.; Adami, D.; Lu, B.; Liu, C.; Maazouz, A. Multiscale Structural Evolution and Its Relationship to Dielectric Properties of Micro-/Nano-Layer Coextruded PVDF-HFP/PC Films. Polymers 2020, 12, 2596. [Google Scholar] [CrossRef] [PubMed]
- Fredrickson, G.H.; Milner, S.T. Time-Dependent Reactive Coupling at Polymer−Polymer Interfaces. Macromolecules 1996, 29, 7386–7390. [Google Scholar] [CrossRef]
- Fredrickson, G.H. Diffusion-controlled reactions at polymer-polymer interfaces. Phys. Rev. Lett. 1996, 76, 3440. [Google Scholar] [CrossRef]
- O’shaughnessy, B.; Sawhney, U. Polymer reaction kinetics at interfaces. Phys. Rev. Lett. 1996, 76, 3444. [Google Scholar] [CrossRef]
- O’Shaughnessy, B.; Sawhney, U. Reaction Kinetics at Polymer−Polymer Interfaces. Macromolecules 1996, 29, 7230–7239. [Google Scholar] [CrossRef]
- Wang, M.; Yuan, G.; Han, C.C. Reaction process in polycarbonate/polyamide bilayer film and blend. Polymer 2013, 54, 3612–3619. [Google Scholar] [CrossRef]
- Lyu, S.-P.; Cernohous, J.J.; Bates, F.S.; Macosko, C.W. Interfacial Reaction Induced Roughening in Polymer Blends. Macromolecules 1999, 32, 106–110. [Google Scholar] [CrossRef]
- Kim, H.Y.; Jeong, U.; Kim, J.K. Reaction Kinetics and Morphological Changes of Reactive Polymer−Polymer Interface. Macromolecules 2003, 36, 1594–1602. [Google Scholar] [CrossRef]
- Kim, H.Y.; Kim, H.J.; Kim, J.K. Effect of interfacial reaction and morphology on rheological properties of reactive bilayer. Polym. J. 2006, 38, 1165–1172. [Google Scholar] [CrossRef] [Green Version]
- Lamnawar, K.; Baudouin, A.; Maazouz, A. Interdiffusion/reaction at the polymer/polymer interface in multilayer systems probed by linear viscoelasticity coupled to FTIR and NMR measurements. Eur. Polym. J. 2010, 46, 1604–1622. [Google Scholar] [CrossRef]
- Lu, B.; Lamnawar, K.; Maazouz, A. Rheological and dynamic insights into an in situ reactive interphase with graft copolymers in multilayered polymer systems. Soft Matter 2017, 13, 2523–2535. [Google Scholar] [CrossRef]
- Kim, H.Y.; Lee, D.H.; Kim, J.K. Effect of oscillatory shear on the interfacial morphology of a reactive bilayer polymer system. Polymer 2006, 47, 5108–5116. [Google Scholar] [CrossRef]
- Kim, H.Y.; Joo, W.; Kim, J.K. Effect of Perpendicular Shear Force on the Interfacial Morphology of Reactive Polymer Bilayer. Macromol. Chem. Phys. 2008, 209, 746–753. [Google Scholar] [CrossRef]
- Zhang, J.; Ji, S.; Song, J.; Lodge, T.P.; Macosko, C.W. Flow Accelerates Interfacial Coupling Reactions. Macromolecules 2010, 43, 7617–7624. [Google Scholar] [CrossRef]
- Kiparissoff-Bondil, H.; Devisme, S.; Rauline, D.; Chopinez, F.; Restagno, F.; Léger, L. Evidences for flow-assisted interfacial reaction in coextruded PA6/PP/PA6 films. Polym. Eng. Sci. 2019, 59, E44–E50. [Google Scholar] [CrossRef]
- Barraud, T.; Devisme, S.; Hervet, H.; Brunello, D.; Klein, V.; Poulard, C.; Restagno, F.; Léger, L. Convection and diffusion assisted reactive coupling at incompatible semi-crystalline polymer interfaces. J. Phys. Mater. 2020, 3, 035001. [Google Scholar] [CrossRef]
- Song, J.; Baker, A.M.; Macosko, C.W.; Ewoldt, R.H. Reactive Coupling between Immiscible Polymer Chains: Acceleration by Compressive Flow. AlChE J. 2013, 59, 3391–3402. [Google Scholar] [CrossRef]
- Du, Q.; Jiang, G.; Li, J.; Guo, S. Adhesion and delamination failure mechanisms in alternating layered polyamide and polyethylene with compatibilizer. Polym. Eng. Sci. 2010, 50, 1111–1121. [Google Scholar] [CrossRef]
- Lamnawar, K.; Maazouz, A. Rheological study of multilayer functionalized polymers: Characterization of interdiffusion and reaction at polymer/polymer interface. Rheol. Acta 2006, 45, 411–424. [Google Scholar] [CrossRef]
- Song, J.; Ewoldt, R.H.; Hu, W.; Craig Silvis, H.; Macosko, C.W. Flow accelerates adhesion between functional polyethylene and polyurethane. AlChE J. 2011, 57, 3496–3506. [Google Scholar] [CrossRef]
- Lu, B.; Lamnawar, K.; Maazouz, A. Influence of in situ reactive interphase with graft copolymer on shear and extensional rheology in a model bilayered polymer system. Polym. Test. 2017, 61, 289–299. [Google Scholar] [CrossRef]
- Bondon, A.; Lamnawar, K.; Maazouz, A. Influence of copolymer architecture on generation of defects in reactive multilayer coextrusion. Key Eng. Mater. 2015, 651, 836–841. [Google Scholar] [CrossRef]
- Bondon, A.; Lamnawar, K.; Maazouz, A. Experimental investigation of a new type of interfacial instability in a reactive coextrusion process. Polym. Eng. Sci. 2015, 55, 2542–2552. [Google Scholar] [CrossRef]
- Lu, B.; Bondon, A.; Touil, I.; Zhang, H.; Alcouffe, P.; Pruvost, S.; Liu, C.; Maazouz, A.; Lamnawar, K. Role of the Macromolecular Architecture of Copolymers at Layer–Layer Interfaces of Multilayered Polymer Films: A Combined Morphological and Rheological Investigation. Ind. Eng. Chem. Res. 2020, 59, 22144–22154. [Google Scholar] [CrossRef]
- Vuong, S.; Léger, L.; Restagno, F. Controlling interfacial instabilities in PP/EVOH coextruded multilayer films through the surface density of interfacial copolymers. Polym. Eng. Sci. 2020, 60, 1420–1429. [Google Scholar] [CrossRef]
- Michell, R.M.; Müller, A.J. Confined crystallization of polymeric materials. Prog. Polym. Sci. 2016, 54 (Suppl. C), 183–213. [Google Scholar] [CrossRef]
- Mijangos, C.; Hernández, R.; Martín, J. A review on the progress of polymer nanostructures with modulated morphologies and properties, using nanoporous AAO templates. Prog. Polym. Sci. 2016, 54 (Suppl. C), 148–182. [Google Scholar] [CrossRef]
- Cangialosi, D.; Alegría, A.; Colmenero, J. Effect of nanostructure on the thermal glass transition and physical aging in polymer materials. Prog. Polym. Sci. 2016, 54 (Suppl. C), 128–147. [Google Scholar] [CrossRef]
- Wang, H.; Keum, J.K.; Hiltner, A.; Baer, E.; Freeman, B.; Rozanski, A.; Galeski, A. Confined Crystallization of Polyethylene Oxide in Nanolayer Assemblies. Science 2009, 323, 757–760. [Google Scholar] [CrossRef] [PubMed]
- Beadie, G.; Shirk, J.S.; Rosenberg, A.; Lane, P.A.; Fleet, E.; Kamdar, A.R.; Jin, Y.; Ponting, M.; Kazmierczak, T.; Yang, Y.; et al. Optical properties of a bio-inspired gradient refractive index polymer lens. Opt. Express 2008, 16, 11540–11547. [Google Scholar] [CrossRef]
- Jin, Y.; Rogunova, M.; Hiltner, A.; Baer, E.; Nowacki, R.; Galeski, A.; Piorkowska, E. Structure of polypropylene crystallized in confined nanolayers. J. Polym. Sci. Part. B Polym. Phys. 2004, 42, 3380–3396. [Google Scholar] [CrossRef]
- Luo, S.; Yi, L.; Zheng, Y.; Shen, J.; Guo, S. Crystallization of polypropylene in multilayered spaces: Controllable morphologies and properties. Eur. Polym. J. 2017, 89, 138–149. [Google Scholar] [CrossRef]
- Bernal-Lara, T.E.; Masirek, R.; Hiltner, A.; Baer, E.; Piorkowska, E.; Galeski, A. Morphology studies of multilayered HDPE/PS systems. J. Appl. Polym. Sci. 2006, 99, 597–612. [Google Scholar] [CrossRef]
- Cheng, J.; Pu, H. Orientation of LDPE crystals from microscale to nanoscale via microlayer or nanolayer coextrusion. Chin. J. Polym. Sci. 2016, 34, 1411–1422. [Google Scholar] [CrossRef]
- Ponting, M.; Lin, Y.; Keum, J.K.; Hiltner, A.; Baer, E. Effect of Substrate on the Isothermal Crystallization Kinetics of Confined Poly(ε-caprolactone) Nanolayers. Macromolecules 2010, 43, 8619–8627. [Google Scholar] [CrossRef]
- Wang, H.; Keum, J.K.; Hiltner, A.; Baer, E. Crystallization Kinetics of Poly(ethylene oxide) in Confined Nanolayers. Macromolecules 2010, 43, 3359–3364. [Google Scholar] [CrossRef]
- Wang, H.; Keum, J.K.; Hiltner, A.; Baer, E. Confined Crystallization of PEO in Nanolayered Films Impacting Structure and Oxygen Permeability. Macromolecules 2009, 42, 7055–7066. [Google Scholar] [CrossRef]
- Chen, X.; Tseng, J.-K.; Treufeld, I.; Mackey, M.; Schuele, D.E.; Li, R.; Fukuto, M.; Baer, E.; Zhu, L. Enhanced Dielectric Properties due to Space Charge-induced Interfacial Polarization in Multilayer Polymer Films. J. Mater. Chem. C 2017, 5, 10417–10426. [Google Scholar] [CrossRef]
- Huang, H.; Chen, X.; Li, R.; Fukuto, M.; Schuele, D.E.; Ponting, M.; Langhe, D.; Baer, E.; Zhu, L. Flat-On Secondary Crystals as Effective Blocks To Reduce Ionic Conduction Loss in Polysulfone/Poly(vinylidene fluoride) Multilayer Dielectric Films. Macromolecules 2018, 51, 5019–5026. [Google Scholar] [CrossRef]
- Mackey, M.; Schuele, D.E.; Zhu, L.; Baer, E. Layer confinement effect on charge migration in polycarbonate/poly(vinylidene fluorid-co-hexafluoropropylene) multilayered films. J. Appl. Phys. 2012, 111, 113702. [Google Scholar] [CrossRef] [Green Version]
- Mackey, M.; Schuele, D.E.; Zhu, L.; Flandin, L.; Wolak, M.A.; Shirk, J.S.; Hiltner, A.; Baer, E. Reduction of Dielectric Hysteresis in Multilayered Films via Nanoconfinement. Macromolecules 2012, 45, 1954–1962. [Google Scholar] [CrossRef]
- Chen, X.; Li, Q.; Langhe, D.; Ponting, M.; Li, R.; Fukuto, M.; Baer, E.; Zhu, L. Achieving Flat-on Primary Crystals by Nanoconfined Crystallization in High-Temperature Polycarbonate/Poly(vinylidene fluoride) Multilayer Films and Its Effect on Dielectric Insulation. ACS Appl. Mater. Interfaces 2020, 12, 44892–44901. [Google Scholar] [CrossRef] [PubMed]
- Langhe, D.S.; Murphy, T.M.; Shaver, A.; LaPorte, C.; Freeman, B.D.; Paul, D.R.; Baer, E. Structural relaxation of polystyrene in nanolayer confinement. Polymer 2012, 53, 1925–1931. [Google Scholar] [CrossRef]
- Arabeche, K.; Delbreilh, L.; Adhikari, R.; Michler, G.H.; Hiltner, A.; Baer, E.; Saiter, J.-M. Study of the Cooperativity at the Glass Transition Temperature in PC/PMMA Multilayered Films: Influence of Thickness Reduction from Macro- to Nanoscale. Polymer 2012, 53, 1355–1361. [Google Scholar] [CrossRef]
- Casalini, R.; Zhu, L.; Baer, E.; Roland, C.M. Segmental Dynamics and the Correlation Length in Nanoconfined PMMA. Polymer 2016, 88, 133–136. [Google Scholar] [CrossRef] [Green Version]












Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lu, B.; Zhang, H.; Maazouz, A.; Lamnawar, K. Interfacial Phenomena in Multi-Micro-/Nanolayered Polymer Coextrusion: A Review of Fundamental and Engineering Aspects. Polymers 2021, 13, 417. https://doi.org/10.3390/polym13030417
Lu B, Zhang H, Maazouz A, Lamnawar K. Interfacial Phenomena in Multi-Micro-/Nanolayered Polymer Coextrusion: A Review of Fundamental and Engineering Aspects. Polymers. 2021; 13(3):417. https://doi.org/10.3390/polym13030417
Chicago/Turabian StyleLu, Bo, Huagui Zhang, Abderrahim Maazouz, and Khalid Lamnawar. 2021. "Interfacial Phenomena in Multi-Micro-/Nanolayered Polymer Coextrusion: A Review of Fundamental and Engineering Aspects" Polymers 13, no. 3: 417. https://doi.org/10.3390/polym13030417