
Chitosan Reinforced with Kenaf Nanocrystalline Cellulose as an Effective Carrier for the Delivery of Platelet Lysate in the Acceleration of Wound Healing



Abstract
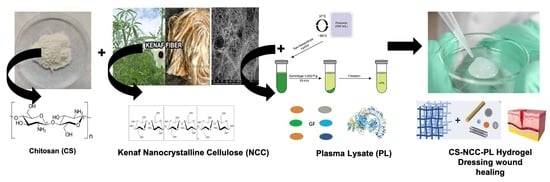
:
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Preparation of Nanocrystalline Cellulose from Kenaf Bast Fibers
2.3. PL Processing and Quantification of GFs
2.4. Preparation of CS Hydrogel and CS-NCC Composite Hydrogels
2.5. Characterization of NCC and Hydrogels
2.5.1. Chemical Composition, Fiber Yield, and Zeta Potential
2.5.2. X-ray Diffraction Characterization (XRD)
2.5.3. Scanning Electron Microscopy (SEM)
2.5.4. Fourier Transform Infrared Spectroscopy-Attenuated Total Reflectance (FTIR-ATR) Spectroscopy
2.5.5. Swelling Behavior of Hydrogels
2.5.6. Moisture Loss Study
2.5.7. In Vitro Protein Release Assessment
2.6. Cytocompatibility Studies
2.6.1. Isolation of Human Dermal Fibroblast (HDF)
2.6.2. Cell Culture
2.6.3. Cell Viability
2.6.4. Cell Proliferation
2.6.5. Scratch Wound Assay
2.6.6. LIVE/DEAD® Cell Viability Assay
2.6.7. CS Stabilizing Effect on PL
Protease Degradation Test
Heat Treatment Test
2.7. Statistical Analysis
3. Results
3.1. Quantification of GFs in PL
3.2. Chemical Composition, Fiber Yield, Zeta Potential, and Crystallinity of NCC
3.3. Scanning Electron Microscopy
3.4. FTIR-ATR Spectroscopic Analysis of NCC
3.5. Swelling Study
3.6. Moisture Loss Study
3.7. In Vitro Protein Release Assay
3.7.1. Cell Viability
3.7.2. Cell Proliferation
3.8. Wound Scratch Assay
3.9. LIVE/DEAD® Cell Viability Assay
3.10. CS Stabilizing Effect on PL
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Tang, J.; Grishkewich, N.; Tam, K.C. Functionalization of cellulose nanocrystals for advanced applications. J. Colloid Int. Sci. 2017, 494, 397–409. [Google Scholar] [CrossRef] [PubMed]
- Sulaiman, S.; Mokhtar, M.N.; Naim, M.N.; Baharuddin, A.S.; Salleh, M.A.M.; Sulaiman, A. Penghasilan nano-gentian selulosa (CNF) diperolehi daripada gentian kulit kenaf dan potensinya sebagai penyokong pemegunan enzim. Malay. J. Anal. Sci. 2016, 20, 309–317. [Google Scholar] [CrossRef]
- Davoudpour, Y.; Hossain, M.S.; Abdul Khalil, H.P.S.; Mohamad Haafiz, M.K.; Mohd Ishak, Z.A.; Hassan, A.; Sarker, Z.I. Optimization of high pressure homogenization parameters for the isolation of cellulosic nanofibers using response surface methodology. Ind. Crop. Prod. 2015, 74, 381–387. [Google Scholar] [CrossRef]
- Law, J.X.; Chowdhury, S.R.; Saim, A.; Idrus, R.B.H. Platelet-rich plasma with keratinocytes and fibroblasts enhance healing of full-thickness wounds. J. Tissue Viability 2017, 26, 208–215. [Google Scholar] [CrossRef]
- Tsai, H.C.; Lehman, C.W.; Chen, C.M. Use of platelet-rich plasma and platelet-derived patches to treat chronic wounds. J. Wound Care 2019, 28, 15–21. [Google Scholar] [CrossRef]
- Han, D.; He, Z.; Zhong, R.; Zhang, X.; Wang, H. Preparation and characterization of a hemostatic porous platelet-rich plasma chitosan/silk fibroin wound dressing. Chin. J. Biotechnol. 2020, 36, 332–340. [Google Scholar] [CrossRef]
- Seeger, A.; Paller, S. The roles of growth factors in keratinocyte migration. Adv. Wound Care 2015, 4, 213–224. [Google Scholar] [CrossRef] [Green Version]
- Stolzenburg-Veeser, L.; Golubnitschaja, O. Mini-encyclopaedia of the wound healing—Opportunities for integrating multi-omic approaches into medical practice. J. Proteom. 2018, 188, 71–84. [Google Scholar] [CrossRef]
- Busilacchi, A.; Gigante, A.; Mattioli-Belmonte, M.; Manzotti, S.; Muzzarelli, R.A.A. Chitosan stabilizes platelet growth factors and modulates stem cell differentiation toward tissue regeneration. Carbohydr. Polym. 2013, 98, 665–676. [Google Scholar] [CrossRef] [PubMed]
- Dai, T.; Tanaka, M.; Huang, Y.Y.; Hamblin, M.R. Chitosan preparations for wounds and burns: Antimicrobial and wound-healing effects. Exp. Rev. Anti-Inf. Ther. 2011, 309, 857–879. [Google Scholar] [CrossRef] [PubMed]
- Dash, M.; Chiellini, F.; Ottenbrite, R.M.; Chiellini, E. Chitosan—A versatile semi-synthetic polymer in biomedical applications. Prog. Polym. Sci. 2011, 36, 981–1014. [Google Scholar] [CrossRef]
- Giri, T.K.; Thakur, A.; Alexander, A.; Ajazuddin; Badwaik, H.; Tripathi, D.K. Modified chitosan hydrogels as drug delivery and tissue engineering systems: Present status and applications. Acta Pharm. Sin. B 2012, 2, 439–449. [Google Scholar] [CrossRef] [Green Version]
- Patrulea, V.; Ostafe, V.; Borchard, G.; Jordan, O. Chitosan as a starting material for wound healing applications. Eur. J. Pharm. Biopharm. 2015, 97, 417–426. [Google Scholar] [CrossRef] [Green Version]
- Rossi, S.; Faccendini, M.C.; Bonferoni, F.; Ferrari, G.; Sandri, C.; Caramella, C.M. “Sponge-like” dressings based on biopolymers for the delivery of platelet lysate to skin chronic wounds. Int. J. Pharm. 2013, 440, 207–215. [Google Scholar] [CrossRef] [PubMed]
- Mohammadi, R.; Mehrtash, M.; Mehrtash, M.; Hassani, N.; Hassanpour, A. effect of platelet rich plasma combined with chitosan biodegradable film on full-thickness wound healing in rat model. Bull. Emerg. Traum. 2016, 4, 29. [Google Scholar]
- Fischer, T.H.; Thatte, H.S.; Nichols, T.C.; Bender-Neal, D.E.; Bellinger, D.A.; Vournakis, J.N. Synergistic platelet integrin signaling and factor XII activation in poly-N-acetyl glucosamine fiber-mediated hemostasis. Biomaterials 2005, 26, 5433–5443. [Google Scholar] [CrossRef]
- Wang, Z.; Lu, W.W.; Zhen, W.; Yang, D.S. Novel biomaterial strategies for controlled growth factor delivery for biomedical applications. NPG Asia Mater. 2017, 9, e435. [Google Scholar] [CrossRef]
- Dorishetty, P.; Balu, R.; Athukoralalage, S.S.; Greaves, T.L.; Mata, J.; de Campo, L.; Saha, N.; Zannettino, A.C.W.; Dutta, N.K.; Choudhury, N.R. Tunable biomimetic hydrogels from silk fibroin and nanocellulose. ACS Sustain. Chem. Eng. 2020, 8, 2375–2389. [Google Scholar] [CrossRef]
- Liu, J.; Cheng, F.; Grénman, H.; Spoljaric, S.; Seppälä, J.; Eriksson, J.E.; Willför, S.; Xu, C. Development of nanocellulose scaffolds with tunable structures to support 3D cell culture. Carbohydr. Polym. 2016, 148, 259–271. [Google Scholar] [CrossRef] [PubMed]
- Song, K.; Zhu, X.; Zhu, W.; Li, X. Preparation and characterization of cellulose nanocrystal extracted from Calotropis procera biomass. Bioresour. Bioprocess. 2019, 45, 1–8. [Google Scholar] [CrossRef] [Green Version]
- Kutlu, B.; Tiǧli Aydin, R.S.; Akman, A.C.; Gümüşderelioglu, M.; Nohutcu, R.M. Platelet-rich plasma-loaded chitosan scaffolds: Preparation and growth factor release kinetics. J. Biomed. Mater. Res. Part B Appl. Biomater. 2013, 101B, 28–35. [Google Scholar] [CrossRef] [PubMed]
- Doench, I.; Tran, T.A.; David, L.; Montembault, A.; Viguier, E.; Gorzelanny, C.G.; Sudre, T.; Cachon, M.; Louback-Mohamed, M.; Horbelt, N.; et al. Cellulose nanofiber-reinforced chitosan hydrogel composites for intervertebral disc tissue repair. Biomimetics 2019, 4, 19. [Google Scholar] [CrossRef] [Green Version]
- Tuerxun, D.; Pulingam, T.; Nordin, N.I.; Chen, Y.W.; Bin Kamaldin, J.; Julkapli, N.B.M.; Lee, H.V.; Leo, B.F.; Bin Johan, M.R. Synthesis, characterization and cytotoxicity studies of nanocrystalline cellulose from the production waste of rubber-wood and kenaf-bast fibers. Eur. Polym. J. 2019, 116, 352–360. [Google Scholar] [CrossRef]
- Sri Aprilia, N.A.; Arahman, N. Properties of nanocrystalline cellulose from pineapple crown leaf waste. In IOP Conference Series: Materials Science and Engineering; IOP Publishing: Bristol, UK, 2020; Volume 796. [Google Scholar] [CrossRef]
- Maache, M.; Bezazi, A.; Amroune, S.; Scarpa, F.; Dufresne, A. Characterization of a novel natural cellulosic fiber from Juncus effusus L. Carbohydr. Polym. 2017, 171, 163–172. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lai, J.C.Y.; Lai, H.Y.; Rao, N.K.; Ng, S.F. Treatment for diabetic ulcer wounds using a fern tannin optimized hydrogel formulation with antibacterial and antioxidative properties. J. Ethnopharmacol. 2016, 189, 277–289. [Google Scholar] [CrossRef]
- Xi Loh, E.Y.; Fauzi, M.B.; Ng, M.H.; Ng, P.Y.; Ng, S.F.; Ariffin, H.; Mohd Amin, M.C.I. Cellular and molecular interaction of human dermal fibroblasts with bacterial nanocellulose composite hydrogel for tissue regeneration. ACS Appl. Mater. Interfaces 2018, 10, 39532–39543. [Google Scholar] [CrossRef]
- Nishimoto, S.; Fujita, K.; Sotsuka, Y.; Kinoshita, M.; Fujiwara, T.; Kawai, K.; Kakibuchi, M. Growth factor measurement and histological analysis in platelet rich fibrin: A pilot study. J. Maxillofac. Oral Surg. 2015, 14, 907–913. [Google Scholar] [CrossRef] [Green Version]
- Jonoobi, M.; Khazaeian, A.; Tahir, P.M.; Azry, S.S.; Oksman, K. Characteristics of cellulose nanofibers isolated from rubberwood and empty fruit bunches of oil palm using chemo-mechanical process. Cellulose 2011, 18, 1085–1095. [Google Scholar] [CrossRef]
- Garcia de Rodriguez, N.L.; Thielemans, W.A. Dufresne, Sisal cellulose whiskers reinforced polyvinyl acetate nanocomposites. Cellulose 2006, 13, 261–270. [Google Scholar] [CrossRef]
- Ilyas, R.A.; Sapuan, S.M.; Ishak, M.R. Isolation and characterization of nanocrystalline cellulose from sugar palm fibres (Arenga Pinnata). Carbohydr. Polym. 2018, 181, 1038–1051. [Google Scholar] [CrossRef] [PubMed]
- Karagkiozaki, V.; Logothetidis, S.; Lousinian, S.; Giannoglo, G. Impact of surface electric properties of carbon-based thin films on platelets activation for nano-medical and nano-sensing applications. Int. J. Nanomed. 2008, 3, 461. [Google Scholar] [CrossRef]
- Karimi, S.; Tahir, P.M.; Karimi, A.; Dufresne, A.; Abdulkhani, A. Kenaf bast cellulosic fibers hierarchy: A comprehensive approach from micro to nano. Carbohydr. Polym. 2014, 101, 878–885. [Google Scholar] [CrossRef] [PubMed]
- Khan, A.; Jawaid, M.; Kia Kian, L.; Khan, A.A.P.; Asiri, A.M. Isolation and production of nanocrystalline cellulose from conocarpus fiber. Polymers 2021, 13, 1835. [Google Scholar] [CrossRef]
- Lv, W.; Xia, Z.; Song, Y.; Wang, P.; Liu, S.; Zhang, Y.; Ben, H.; Han, G.; Jiang, W. Using microwave assisted organic acid treatment to separate cellulose fiber and lignin from kenaf bast. Ind. Crop. Prod. 2021, 171, 113934. [Google Scholar] [CrossRef]
- Le Troedec, M.; Sedan, D.; Peyratout, C.; Bonnet, J.P.; Smith, A.; Guinebretiere, R.; Gloaguen, V.; Krausz, P. Influence of various chemical treatments on the composition and structure of hemp fibres. Compos. Part A Appl. Sci. Manuf. 2008, 39, 514–522. [Google Scholar] [CrossRef]
- Nacos, M.K.; Katapodis, P.; Pappas, C.; Daferera, D.; Tarantilis, P.A.; Christakopoulos, P.; Polissiou, M. Kenaf xylan—A source of biologically active acidic oligosaccharides. Carbohydr. Polym. 2006, 66, 126–134. [Google Scholar] [CrossRef]
- Tang, Y.; Zhang, H.; Wei, Q.; Tang, X.; Zhuang, W. Biocompatible chitosan-collagen-hydroxyapatite nanofibers coated with platelet-rich plasma for regenerative engineering of the rotator cuff of the shoulder. RSC Adv. 2019, 9, 27013–27020. [Google Scholar] [CrossRef] [Green Version]
- Song, W.; Xu, J.; Gao, L.; Zhang, Q.; Tong, J. Preparation of Freeze-Dried Porous Chitosan Microspheres for the Removal of Hexavalent Chromium. Appl. Sci. 2021, 11, 4217. [Google Scholar] [CrossRef]
- Sampath, U.G.T.M.; Ching, Y.C.; Chuah, C.H.; Singh, R.; Lin, P.-C. Preparation and characterization of nanocellulose reinforced semi-interpenetrating polymer network of chitosan hydrogel. Cellulose 2017, 24, 2215–2228. [Google Scholar] [CrossRef]
- Hattori, H.; Ishihara, M. Feasibility of improving platelet-rich plasma therapy by using chitosan with high platelet activation ability. Exp. Ther. Med. 2017, 13, 1176–1180. [Google Scholar] [CrossRef] [Green Version]















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Material | Chemical Composition (%) | ||
|---|---|---|---|
| Cellulose | Hemicellulose | Lignin | |
| Raw | 30 | 31 | 30 |
| Alkali-Treated | 70 | 20 | 15 |
| Bleached | 84 | 5 | 3 |
| Raw Fibers Peak (cm−1) | Bleached Fibers Peak (cm−1) | NCC Peak (cm−1) | Peak Assignment |
|---|---|---|---|
| 3345.74 | 3342.62 | 3322.16 | O–H stretching |
| 2909.88 | 2911.71 | 2909.90 | C–H symmetrical stretching and CH2 symmetrical stretching |
| 1326.28 | - | - | C–O aromatic ring |
| 1242.47 | - | - | C–O stretching |
| 1601.19 | - | - | C=C groups |
| - | - | 1634 | Adsorbed water |
| 1028.96 | 1023.74 | 1030.10 | C-O and O-H groups stretching |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bhatnagar, P.; Law, J.X.; Ng, S.-F. Chitosan Reinforced with Kenaf Nanocrystalline Cellulose as an Effective Carrier for the Delivery of Platelet Lysate in the Acceleration of Wound Healing. Polymers 2021, 13, 4392. https://doi.org/10.3390/polym13244392
Bhatnagar P, Law JX, Ng S-F. Chitosan Reinforced with Kenaf Nanocrystalline Cellulose as an Effective Carrier for the Delivery of Platelet Lysate in the Acceleration of Wound Healing. Polymers. 2021; 13(24):4392. https://doi.org/10.3390/polym13244392
Chicago/Turabian StyleBhatnagar, Payal, Jia Xian Law, and Shiow-Fern Ng. 2021. "Chitosan Reinforced with Kenaf Nanocrystalline Cellulose as an Effective Carrier for the Delivery of Platelet Lysate in the Acceleration of Wound Healing" Polymers 13, no. 24: 4392. https://doi.org/10.3390/polym13244392