
Self-Healable and Recyclable Biomass-Derived Polyurethane Networks through Carbon Dioxide Immobilization


Abstract
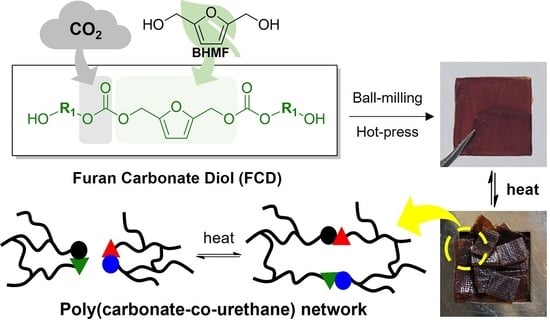
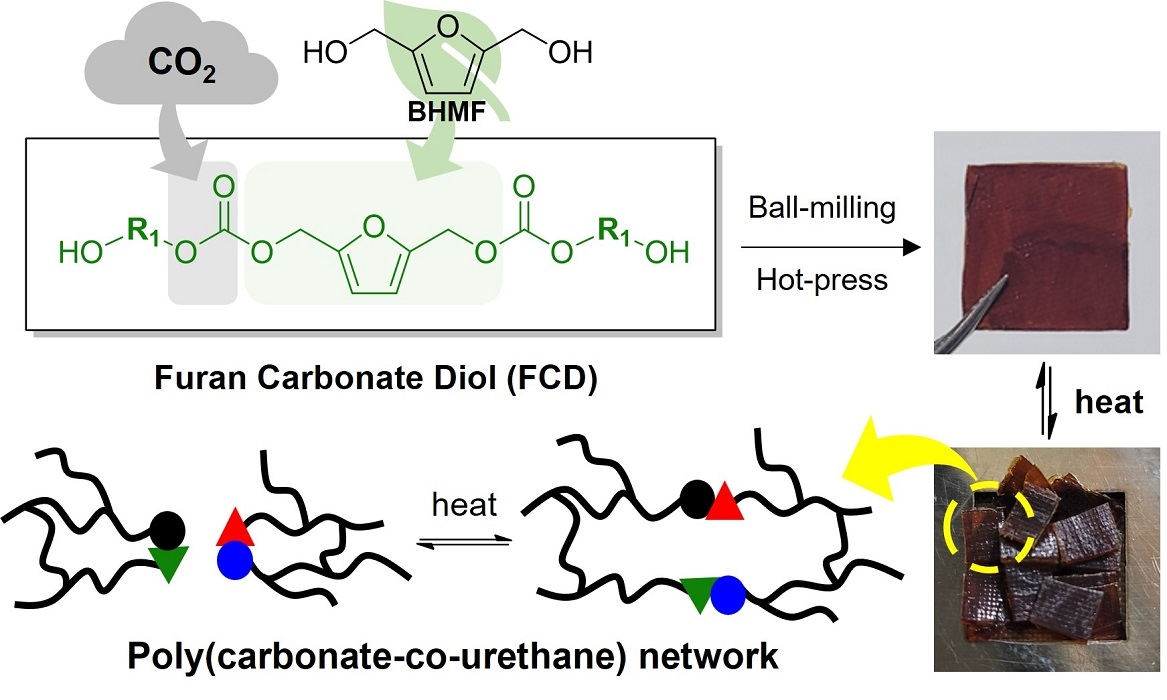
:
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Methods
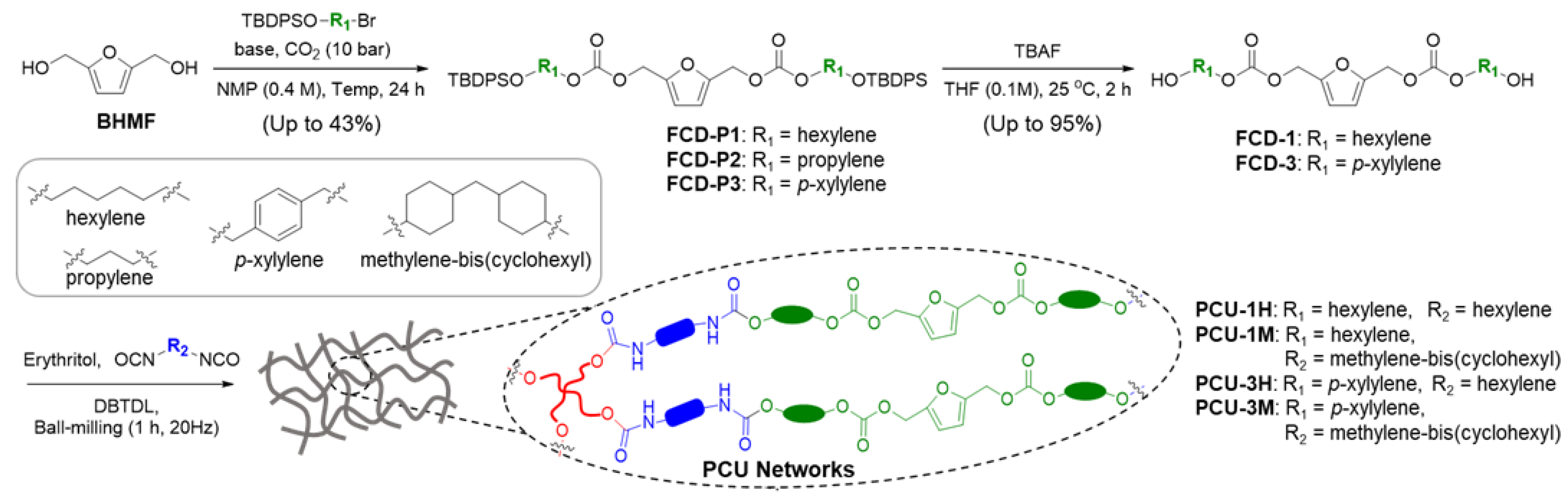
2.2.1. Synthesis of Furan Carbonate Diols Protected with Tert-Butyldiphenylsilane (FCD-Ps)
2.2.2. Synthesis of the Furan Carbonate Diols (FCDs)
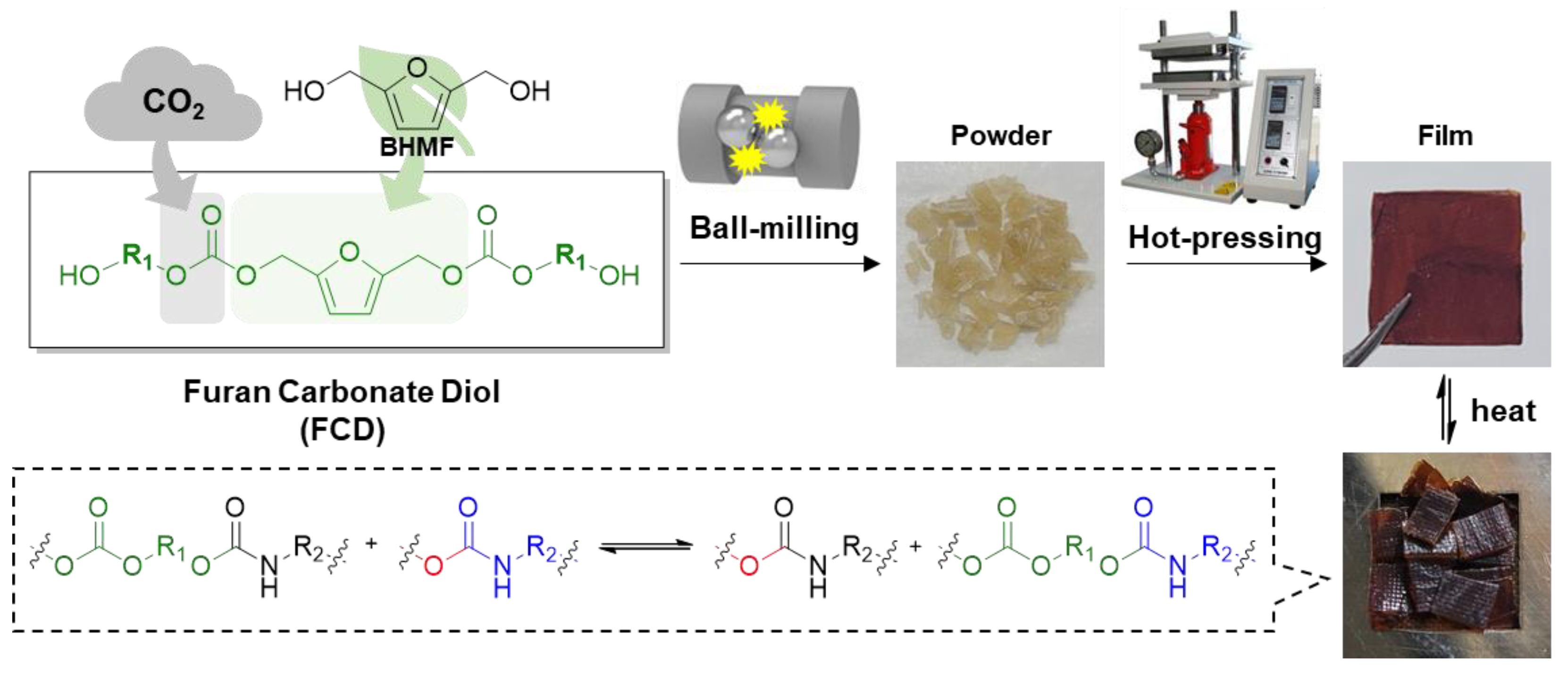
2.2.3. Synthesis of the Poly(Carbonate-co-Urethane) Networks (PCUs) via Ball-Milling
2.2.4. Fabrication of the PCU Films
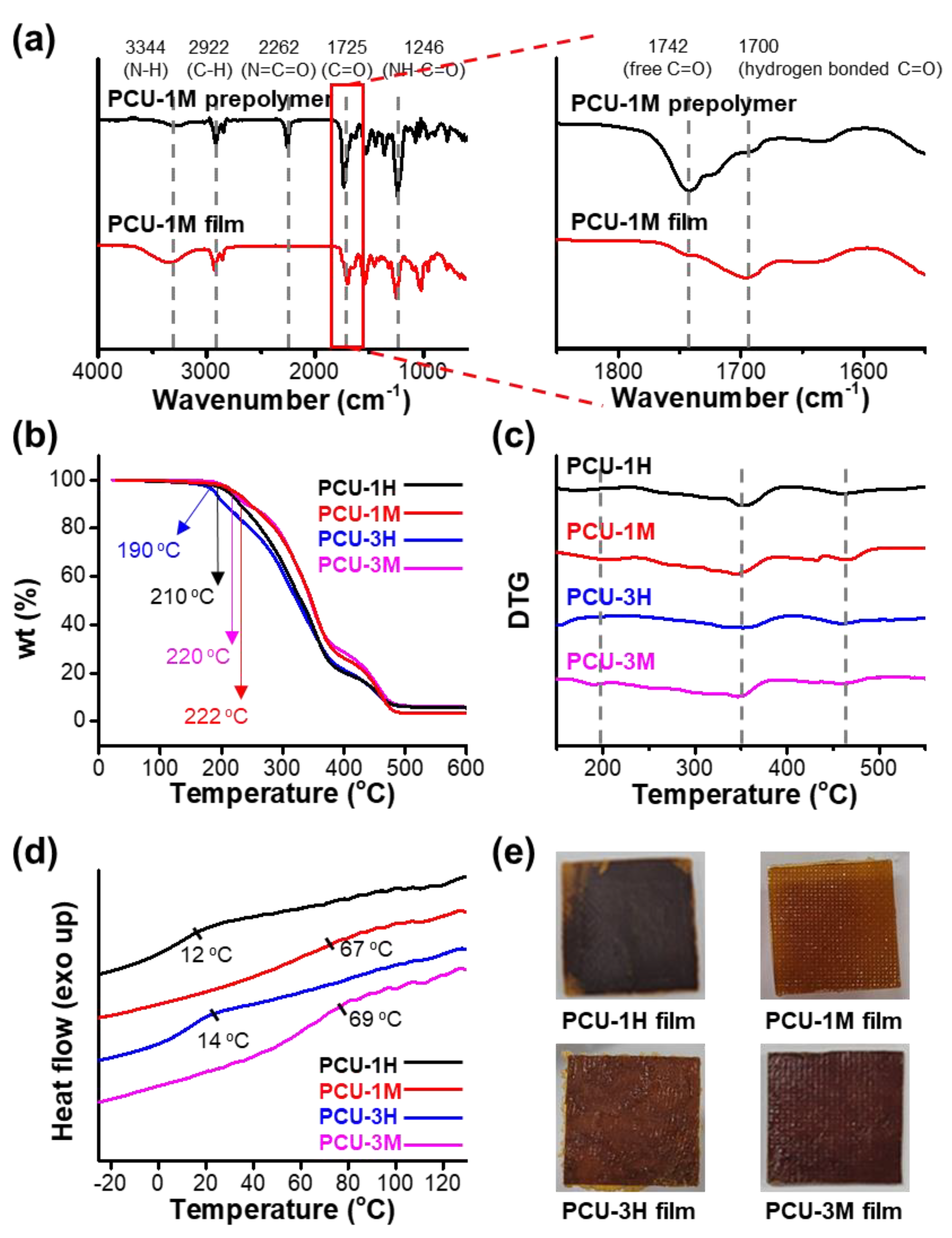
2.3. Measurements
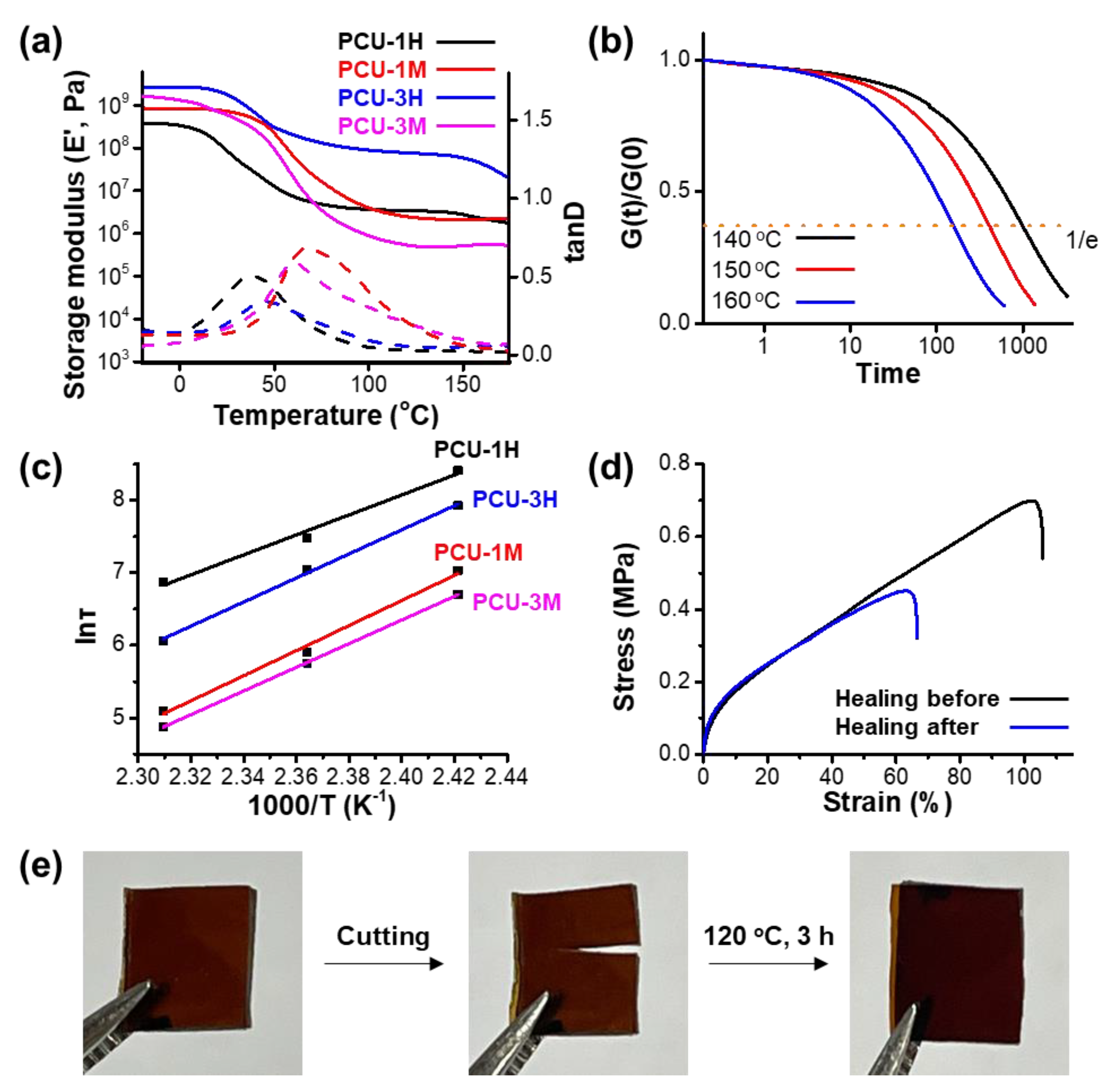
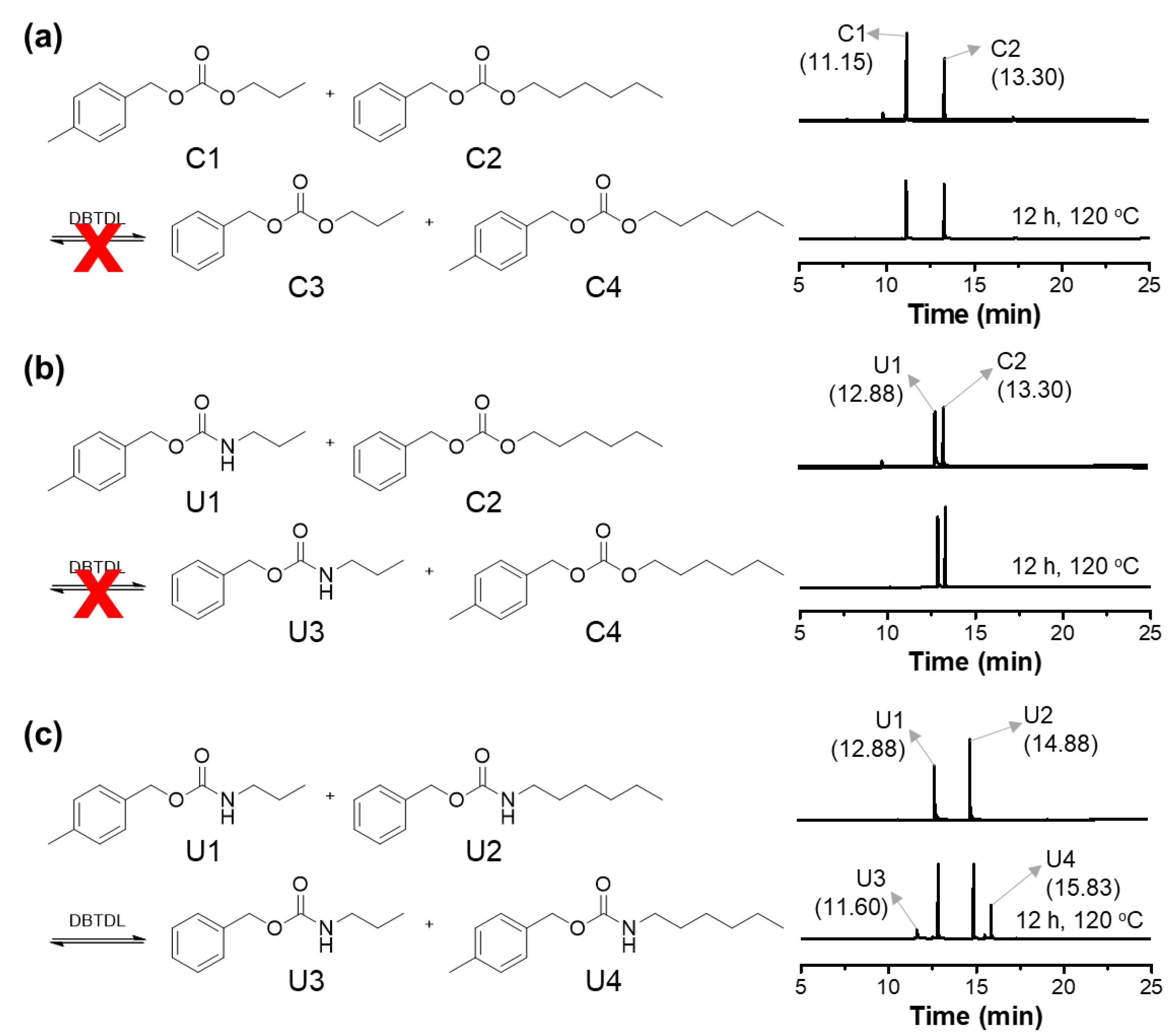
3. Results
Synthesis of CO2-Immobilized Platform FCDs
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Sanz-Pérez, E.S.; Murdock, C.R.; Didas, S.A.; Jones, C.W. Direct capture of CO2 from ambient air. Chem. Rev. 2016, 116, 11840–11876. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Hadjichristidis, N.; Feng, X.; Gnanou, Y. Cs2 CO3-promoted polycondensation of CO2 with diols and dihalides for the synthesis of miscellaneous polycarbonates. Polym. Chem. 2016, 7, 4944–4952. [Google Scholar] [CrossRef]
- Zhou, Q.; Gu, L.; Gao, Y.; Qin, Y.; Wang, X.; Wang, F. Biodegradable CO2-based polycarbonates with rapid and reversible thermal response at body temperature. J. Polym. Sci. Part A Polym. Chem. 2013, 51, 1893–1898. [Google Scholar] [CrossRef]
- Kemper, J. Biomass and carbon dioxide capture and storage: A review. Int. J. Greenh. Gas Control 2015, 40, 401–430. [Google Scholar] [CrossRef]
- Fang, M.; Yi, N.; Di, W.; Wang, T.; Wang, Q. Emission and control of flue gas pollutants in CO2 chemical absorption system–A review. Int. J. Greenh. Gas Control 2020, 93, 102904. [Google Scholar] [CrossRef]
- Zhang, F.; Liu, P.; Zhang, K.; Song, Q.-W. Chemical Adsorption Strategy for DMC-MeOH Mixture Separation. Molecules 2021, 26, 1735. [Google Scholar] [CrossRef]
- Gui, X.; Tang, Z.; Fei, W. CO2 capture with physical solvent dimethyl carbonate at high pressures. J. Chem. Eng. Data 2010, 55, 3736–3741. [Google Scholar] [CrossRef]
- Grignard, B.; Gennen, S.; Jérôme, C.; Kleij, A.W.; Detrembleur, C. Advances in the use of CO2 as a renewable feedstock for the synthesis of polymers. Chem. Soc. Rev. 2019, 48, 4466–4514. [Google Scholar] [CrossRef]
- Hosseinian, A.; Farshbaf, S.; Mohammadi, R.; Monfared, A.; Vessally, E. Advancements in six-membered cyclic carbonate (1, 3-dioxan-2-one) synthesis utilizing carbon dioxide as a C1 source. RSC Adv. 2018, 8, 17976–17988. [Google Scholar] [CrossRef] [Green Version]
- Tamura, M.; Ito, K.; Honda, M.; Nakagawa, Y.; Sugimoto, H.; Tomishige, K. Direct copolymerization of CO2 and diols. Sci. Rep. 2016, 6, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Gregory, G.L.; Ulmann, M.; Buchard, A. Synthesis of 6-membered cyclic carbonates from 1, 3-diols and low CO2 pressure: A novel mild strategy to replace phosgene reagents. Rsc Adv. 2015, 5, 39404–39408. [Google Scholar] [CrossRef] [Green Version]
- Guo, L.; Zhang, R.; Xiong, Y.; Chang, D.; Zhao, H.; Zhang, W.; Zheng, W.; Chen, J.; Wu, X. The Application of Biomass-Based Catalytic Materials in the Synthesis of Cyclic Carbonates from CO2 and Epoxides. Molecules 2020, 25, 3627. [Google Scholar] [CrossRef] [PubMed]
- Thorat, S.D.; Phillips, P.J.; Semenov, V.; Gakh, A. Physical properties of aliphatic polycarbonates made from CO2 and epoxides. J. Appl. Polym. Sci. 2003, 89, 1163–1176. [Google Scholar] [CrossRef]
- Coates, G.W.; Moore, D.R. Discrete metal-based catalysts for the copolymerization of CO2 and epoxides: Discovery, reactivity, optimization, and mechanism. Angew. Chem. Int. Ed. 2004, 43, 6618–6639. [Google Scholar] [CrossRef]
- Darensbourg, D.J.; Wilson, S.J. What’s new with CO2? Recent advances in its copolymerization with oxiranes. Green Chem. 2012, 14, 2665–2671. [Google Scholar] [CrossRef]
- Tiwari, A.; Titinchi, S. Advanced Catalytic Materials; John Wiley & Sons: Hoboken, NJ, USA, 2015. [Google Scholar]
- Paul, S.; Zhu, Y.; Romain, C.; Brooks, R.; Saini, P.K.; Williams, C.K. Ring-opening copolymerization (ROCOP): Synthesis and properties of polyesters and polycarbonates. Chem. Commun. 2015, 51, 6459–6479. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Trott, G.; Saini, P.; Williams, C. Catalysts for CO2/epoxide ring-opening copolymerization. Philos. Trans. R. Soc. A Math. Phys. Eng. Sci. 2016, 374, 20150085. [Google Scholar] [CrossRef] [Green Version]
- Taherimehr, M.; Pescarmona, P.P. Green polycarbonates prepared by the copolymerization of CO2 with epoxides. J. Appl. Polym. Sci. 2014, 131, 41141. [Google Scholar] [CrossRef]
- Stille, J.K. Step-Growth Polymerization. J. Chem. Educ. 1981, 58, 862–866. [Google Scholar] [CrossRef] [Green Version]
- Bian, S.; Pagan, C.; Andrianova “Artemyeva”, A.A.; Du, G. Synthesis of polycarbonates and poly (ether carbonate) s directly from carbon dioxide and diols promoted by a Cs2CO3/CH2Cl2 system. ACS Omega 2016, 1, 1049–1057. [Google Scholar] [CrossRef]
- Kadokawa, J.i.; Habu, H.; Fukamachi, S.; Karasu, M.; Tagaya, H.; Chiba, K. Direct polycondensation of carbon dioxide with xylylene glycols: A new method for the synthesis of polycarbonates. Macromol. Rapid Commun. 1998, 19, 657–660. [Google Scholar] [CrossRef]
- Nettles, J.; Birks, P.; Sucre, E.; Bilby, R. Sustainable production of bioenergy feedstock from the industrial forest: Potential and challenges of operational scale implementation. Curr. Sustain./Renew. Energy Rep. 2015, 2, 121–127. [Google Scholar] [CrossRef] [Green Version]
- Sacia, E.R.; Balakrishnan, M.; Bell, A.T. Biomass conversion to diesel via the etherification of furanyl alcohols catalyzed by Amberlyst-15. J. Catal. 2014, 313, 70–79. [Google Scholar] [CrossRef] [Green Version]
- Dhillon, R.; von Wuehlisch, G. Mitigation of global warming through renewable biomass. Biomass Bioenergy 2013, 48, 75–89. [Google Scholar] [CrossRef]
- de Haro, J.C.; Allegretti, C.; Smit, A.T.; Turri, S.; D’Arrigo, P.; Griffini, G. Biobased polyurethane coatings with high biomass content: Tailored properties by lignin selection. ACS Sustain. Chem. Eng. 2019, 7, 11700–11711. [Google Scholar] [CrossRef]
- Xue, Z.; Ma, M.-G.; Li, Z.; Mu, T. Advances in the conversion of glucose and cellulose to 5-hydroxymethylfurfural over heterogeneous catalysts. RSC Adv. 2016, 6, 98874–98892. [Google Scholar] [CrossRef]
- Lee, C.H.; Takagi, H.; Okamoto, H.; Kato, M. Improving the mechanical properties of isosorbide copolycarbonates by varying the ratio of comonomers. J. Appl. Polym. Sci. 2013, 127, 530–534. [Google Scholar] [CrossRef]
- Maniar, D.; Jiang, Y.; Woortman, A.J.; van Dijken, J.; Loos, K. Furan-Based Copolyesters from Renewable Resources: Enzymatic Synthesis and Properties. ChemSusChem 2019, 12, 990. [Google Scholar] [CrossRef] [Green Version]
- Zaldivar, J.; Nielsen, J.; Olsson, L. Fuel ethanol production from lignocellulose: A challenge for metabolic engineering and process integration. Appl. Microbiol. Biotechnol. 2001, 56, 17–34. [Google Scholar] [CrossRef]
- Awuchi, C.G.; Echeta, K.C. Current developments in sugar alcohols: Chemistry, nutrition, and health concerns of sorbitol, xylitol, glycerol, arabitol, inositol, maltitol, and lactitol. Int. J. Adv. Acad. Res. 2019, 5, 1–33. [Google Scholar]
- Kohli, K.; Prajapati, R.; Sharma, B.K. Bio-based chemicals from renewable biomass for integrated biorefineries. Energies 2019, 12, 233. [Google Scholar] [CrossRef] [Green Version]
- Jeya, M.; Lee, K.-M.; Tiwari, M.K.; Kim, J.-S.; Gunasekaran, P.; Kim, S.-Y.; Kim, I.-W.; Lee, J.-K. Isolation of a novel high erythritol-producing Pseudozyma tsukubaensis and scale-up of erythritol fermentation to industrial level. Appl. Microbiol. Biotechnol. 2009, 83, 225–231. [Google Scholar] [CrossRef] [PubMed]
- Hao, Q.; Li, F.; Li, Q.; Li, Y.; Jia, L.; Yang, J.; Fang, Q.; Cao, A. Preparation and crystallization kinetics of new structurally well-defined star-shaped biodegradable poly (L-lactide) s initiated with diverse natural sugar alcohols. Biomacromolecules 2005, 6, 2236–2247. [Google Scholar] [CrossRef]
- Zhang, S.; Liu, T.; Hao, C.; Wang, L.; Han, J.; Liu, H.; Zhang, J. Preparation of a lignin-based vitrimer material and its potential use for recoverable adhesives. Green Chem. 2018, 20, 2995–3000. [Google Scholar] [CrossRef]
- Schmidt, S.; Gatti, F.J.; Luitz, M.; Ritter, B.S.; Bruchmann, B.; Mülhaupt, R. Erythritol dicarbonate as intermediate for solvent-and isocyanate-free tailoring of bio-based polyhydroxyurethane thermoplastics and thermoplastic elastomers. Macromolecules 2017, 50, 2296–2303. [Google Scholar] [CrossRef]
- Fridrihsone, A.; Stirna, U.; Lazdiņa, B.; Misāne, M.; Vilsone, D. Characterization of polyurethane networks structure and properties based on rapeseed oil derived polyol. Eur. Polym. J. 2013, 49, 1204–1214. [Google Scholar] [CrossRef]
- Kausar, A. Polyurethane/Epoxy Interpenetrating Polymer Network; InTech: London, UK, 2017; Volume 10, pp. 1–16. [Google Scholar]
- Zhang, Y.; Heath, R.; Hourston, D. Morphology, mechanical properties, and thermal stability of polyurethane–epoxide resin interpenetrating polymer network rigid foams. J. Appl. Polym. Sci. 2000, 75, 406–416. [Google Scholar] [CrossRef]
- Atiqah, A.; Mastura, M.T.; Ahmed Ali, B.A.; Jawaid, M.; Sapuan, S.M. A review on polyurethane and its polymer composites. Curr. Org. Synth. 2017, 14, 233–248. [Google Scholar] [CrossRef]
- Yang, X.; Guo, L.; Xu, X.; Shang, S.; Liu, H. A fully bio-based epoxy vitrimer: Self-healing, triple-shape memory and reprocessing triggered by dynamic covalent bond exchange. Mater. Des. 2020, 186, 108248. [Google Scholar] [CrossRef]
- Altuna, F.I.; Pettarin, V.; Williams, R.J. Self-healable polymer networks based on the cross-linking of epoxidised soybean oil by an aqueous citric acid solution. Green Chem. 2013, 15, 3360–3366. [Google Scholar] [CrossRef]
- Li, M.; Ding, H.; Yang, X.; Xu, L.; Xia, J.; Li, S. Preparation and properties of self-healing polyurethane elastomer derived from tung-oil-based polyphenol. ACS Omega 2019, 5, 529–536. [Google Scholar] [CrossRef] [PubMed]
- Sheppard, D.T.; Jin, K.; Hamachi, L.S.; Dean, W.; Fortman, D.J.; Ellison, C.J.; Dichtel, W.R. Reprocessing postconsumer polyurethane foam using carbamate exchange catalysis and twin-screw extrusion. ACS Cent. Sci. 2020, 6, 921–927. [Google Scholar] [CrossRef]
- Erice, A.; de Luzuriaga, A.R.; Matxain, J.M.; Ruipérez, F.; Asua, J.M.; Grande, H.-J.; Rekondo, A. Reprocessable and recyclable crosslinked poly (urea-urethane) s based on dynamic amine/urea exchange. Polymer 2018, 145, 127–136. [Google Scholar] [CrossRef]
- Denissen, W.; Winne, J.M.; Du Prez, F.E. Vitrimers: Permanent organic networks with glass-like fluidity. Chem. Sci. 2016, 7, 30–38. [Google Scholar] [CrossRef] [Green Version]
- Alraddadi, M.A.; Chiaradia, V.; Stubbs, C.J.; Worch, J.C.; Dove, A.P. Renewable and recyclable covalent adaptable networks based on bio-derived lipoic acid. Polym. Chem. 2021, 12, 5796–5802. [Google Scholar] [CrossRef]
- Khan, A.; Ahmed, N.; Rabnawaz, M. Covalent Adaptable Network and Self-Healing Materials: Current Trends and Future Prospects in Sustainability. Polymers 2020, 12, 2027. [Google Scholar] [CrossRef] [PubMed]
- Lamm, M.E.; Song, L.; Wang, Z.; Lamm, B.; Fu, L.; Tang, C. A facile approach to thermomechanically enhanced fatty acid-containing bioplastics using metal–ligand coordination. Polym. Chem. 2019, 10, 6570–6579. [Google Scholar] [CrossRef]
- Hu, J.; Mo, R.; Sheng, X.; Zhang, X. A self-healing polyurethane elastomer with excellent mechanical properties based on phase-locked dynamic imine bonds. Polym. Chem. 2020, 11, 2585–2594. [Google Scholar] [CrossRef]
- Choi, E.H.; Lee, J.; Son, S.U.; Song, C. Biomass-derived furanic polycarbonates: Mild synthesis and control of the glass transition temperature. J. Polym. Sci. Part A Polym. Chem. 2019, 57, 1796–1800. [Google Scholar] [CrossRef]
- Oh, C.; Choi, E.H.; Choi, E.J.; Premkumar, T.; Song, C. Facile Solid-State Mechanochemical Synthesis of Eco-Friendly Thermoplastic Polyurethanes and Copolymers Using a Biomass-Derived Furan Diol. ACS Sustain. Chem. Eng. 2020, 8, 4400–4406. [Google Scholar] [CrossRef]
- Kim, H.; Cha, I.; Yoon, Y.; Cha, B.J.; Yang, J.; Kim, Y.D.; Song, C. Facile Mechanochemical Synthesis of Malleable Biomass-Derived Network Polyurethanes and Their Shape-Memory Applications. ACS Sustain. Chem. Eng. 2021, 9, 6952–6961. [Google Scholar] [CrossRef]
- Oi, S.; Nemoto, K.; Matsuno, S.; Inoue, Y. Direct synthesis of polycarbonates from CO2, diols, and dihalides. Macromol. Rapid Commun. 1994, 15, 133–137. [Google Scholar] [CrossRef]
- Cao, H.; Qi, F.; Liu, R.; Wang, F.; Zhang, C.; Zhang, X.; Chai, Y.; Zhai, L. The influence of hydrogen bonding on N-methyldiethanolamine-extended polyurethane solid–solid phase change materials for energy storage. Rsc Adv. 2017, 7, 11244–11252. [Google Scholar] [CrossRef] [Green Version]
- Jiang, L.; Ren, Z.; Zhao, W.; Liu, W.; Liu, H.; Zhu, C. Synthesis and structure/properties characterizations of four polyurethane model hard segments. R. Soc. Open Sci. 2018, 5, 180536. [Google Scholar] [CrossRef] [Green Version]
- Wright, K.J.; Badwaik, V.D.; Samaddar, S.; Hyun, S.-H.; Glauninger, K.; Eom, T.; Thompson, D.H. Organocatalytic synthesis and evaluation of polycarbonate pendant polymer: β-Cyclodextrin-based nucleic acid delivery vectors. Macromolecules 2018, 51, 670–678. [Google Scholar] [CrossRef]
- Schoustra, S.K.; Dijksman, J.A.; Zuilhof, H.; Smulders, M.M. Molecular control over vitrimer-like mechanics–tuneable dynamic motifs based on the Hammett equation in polyimine materials. Chem. Sci. 2021, 12, 293–302. [Google Scholar] [CrossRef]
- Tellers, J.; Pinalli, R.; Soliman, M.; Vachon, J.; Dalcanale, E. Reprocessable vinylogous urethane cross-linked polyethylene via reactive extrusion. Polym. Chem. 2019, 10, 5534–5542. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Entry | R1 | Base | Temp. | Yield 2 |
|---|---|---|---|---|
| 1 | hexylene (2 eq) | DBU | 80 °C | No reaction |
| 2 | hexylene (2 eq) | K2CO3 | 80 °C | 20% 3 |
| 3 | hexylene (2 eq) | Cs2CO3 | 80 °C | 17% |
| 4 | hexylene (2.2 eq) | Cs2CO3 | 80 °C | 25% (29% 4) |
| 5 | hexylene (2.2 eq) | Cs2CO3 | 100 °C | 43% |
| 6 | propylene (2.2 eq) | Cs2CO3 | 100 °C | 53% |
| 7 | p-xylylene (2.2 eq) | Cs2CO3 | 100 °C | 8% |
| Entry | Alcohol (R1) | Isocyanate (R2) | Tg 2 (°C) | Tg 3 (°C) | Td5% 4 (°C) | Tdmax 5 (°C) | tan δ | E′ 6 (MPa) | Tv 7 (°C) |
|---|---|---|---|---|---|---|---|---|---|
| PCU-1H | FCD-1 | HDI (H) | 38 | 12 | 210 | 490 | 0.35 | 4.2 | 104 |
| PCU 1M | FCD-1 | HMDI (M) | 68 | 67 | 222 | 490 | 0.70 | 2.4 | 72 |
| PCU-3H | FCD-3 | HDI (H) | 46 | 14 | 190 | 490 | 0.52 | 122 | 118 |
| PCU-3M | FCD-3 | HMDI (M) | 60 | 69 | 220 | 490 | 0.60 | 0.54 | 65 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Baek, S.; Lee, J.; Kim, H.; Cha, I.; Song, C. Self-Healable and Recyclable Biomass-Derived Polyurethane Networks through Carbon Dioxide Immobilization. Polymers 2021, 13, 4381. https://doi.org/10.3390/polym13244381
Baek S, Lee J, Kim H, Cha I, Song C. Self-Healable and Recyclable Biomass-Derived Polyurethane Networks through Carbon Dioxide Immobilization. Polymers. 2021; 13(24):4381. https://doi.org/10.3390/polym13244381
Chicago/Turabian StyleBaek, Seohyun, Juhyen Lee, Hyunwoo Kim, Inhwan Cha, and Changsik Song. 2021. "Self-Healable and Recyclable Biomass-Derived Polyurethane Networks through Carbon Dioxide Immobilization" Polymers 13, no. 24: 4381. https://doi.org/10.3390/polym13244381