
Synthesis of Biobased and Hybrid Polyurethane Xerogels from Bacterial Polyester for Potential Biomedical Applications



Abstract
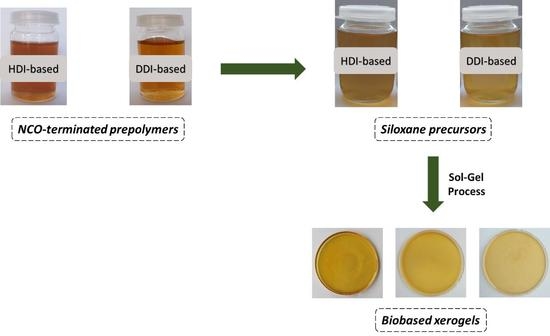
:
1. Introduction
2. Experimental Section
2.1. Materials
2.2. OligoPHB-Diol Synthesis
2.3. Synthesis of PUPs
2.4. Synthesis of SPUs
2.5. Synthesis of Xerogel from SPU
- (1)
- A variation of NCO/OH molar ratio in the PUP (2/1, 4/1 and 6/1), with Rw = 2, giving three xerogels–polymer hybrids named XP2-1R2, XP4-1R2 and XP6-1R2, respectively. A higher NCO/OH molar ratio yield SPU with greater APTMS amount and thus xerogels with higher inorganic networks.
- (2)
- A variation of Rw (1/1, 2/1 and 8/1). This series was prepared from SPU2-1 and the hybrid materials were called XP2-1R1, XP2-1R2 and XP2-1R8. Reducing Rw, hence increasing the water content, decreases the condensations, but promotes hydrolysis reactions in the sol–gel process. However, is it supposed that Rw = 1/1 leads to almost or complete hydrolysis and thus very low or no condensation [27].
- (3)
- The variation of the diisocyanate used in the SPU synthesis: HDI for SPU2-1 and DDI for SPUD2-1, with Rw = 2, giving two different xerogels named XP2-1R2 and XPD2-1R2, respectively. Contrary to HDI, DDI is composed of long aliphatic pending chains, which could bring more mobility and flexibility to the final xerogel.
- (4)
- The gradual replacement of SPU by TEOS using TEOS/SPU ratio of 0, 1/3 and 1/1 and Rw = 2. The corresponding xerogels were named XP2-1R2, XPT3-1 and XPT1-1, respectively. Contrary to APTMS, which has an organic chain and amine end-group chemically linked to the xerogels PU, the incorporation of fully inorganic TEOS promotes hyperbranched Si-O-Si networks with no interaction to the PU.
2.6. General Methods and Analysis
3. Results and Discussion
3.1. Xerogels Synthesis
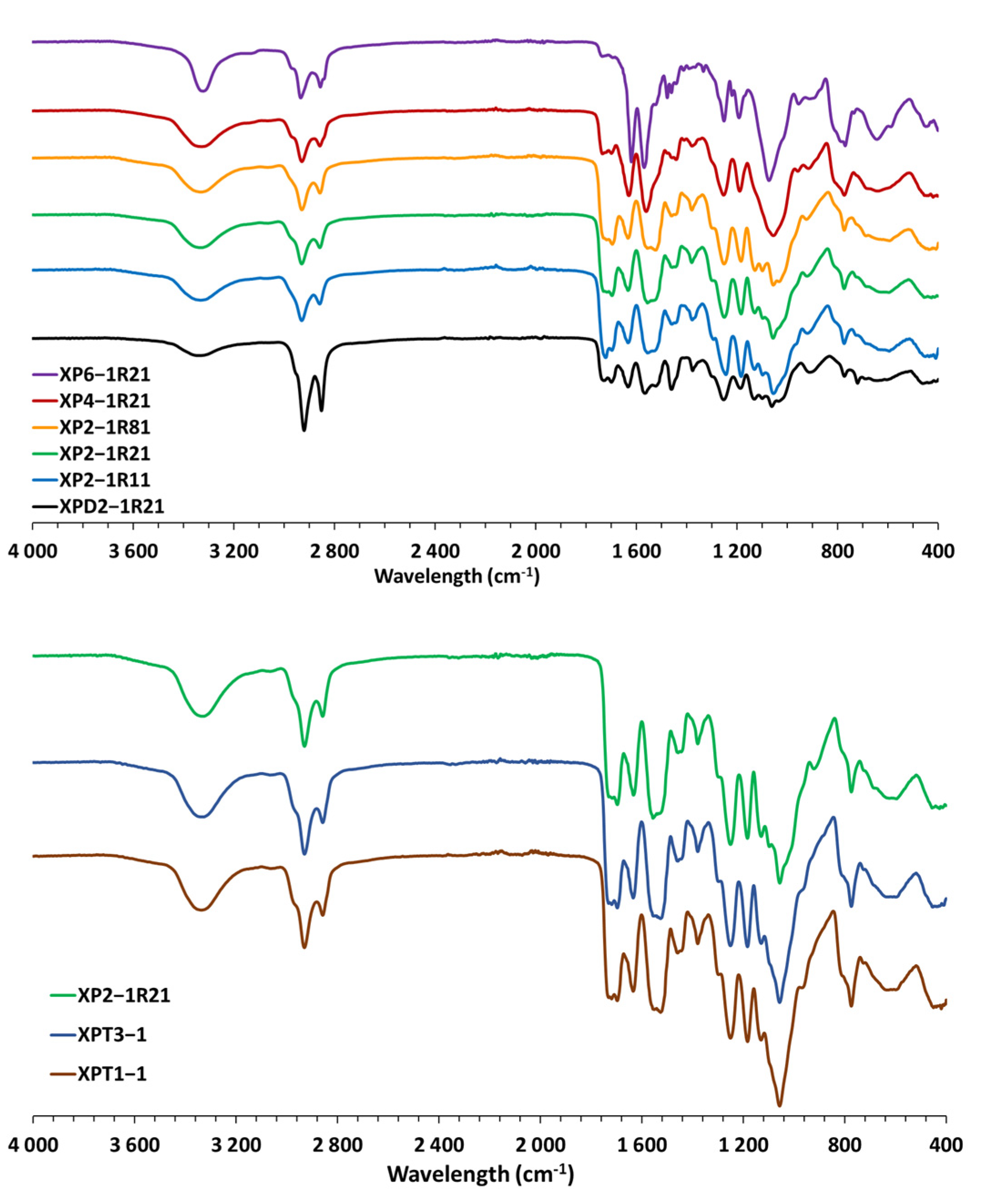
3.2. Analysis of the Hybrid Organic-Inorganic Xerogels
- (1)
- The samples strips were eroded, resulting in flatter surfaces compared to the controls (in Series 2 and more specifically for XP2-1R8);
- (2)
- The samples were eroded between the strips, resulting in more visible strips compared to the control (for XP6-1R2);
- (3)
- The apparition of perforations along the surface (Series 2).
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Wang, X.; Zhang, Y.; Liang, H.; Zhou, X.; Fang, C.; Zhang, C.; Luo, Y. Synthesis and properties of castor oil-based waterborne polyurethane/sodium alginate composites with tunable properties. Carbohydr. Polym. 2019, 208, 391–397. [Google Scholar] [CrossRef]
- Khanderay, J.C.; Gite, V.V. Fully biobased polyester polyols derived from renewable resources toward preparation of polyurethane and their application for coatings. J. Appl. Polym. Sci. 2019, 136, 47558. [Google Scholar] [CrossRef]
- Liao, Z.; Yao, X.; Zhang, L.; Hossain, M.; Wang, J.; Zang, S. Temperature and strain rate dependent large tensile deformation and tensile failure behavior of transparent polyurethane at intermediate strain rates. Int. J. Impact. Eng. 2019, 129, 152–167. [Google Scholar] [CrossRef] [Green Version]
- Wilén, C.-E. The Synthesis of low-viscosity organotin-free moisture-curable silane-terminated Poly(Urethane-Urea)s. Polymers 2018, 10, 781. [Google Scholar]
- Wendels, S.; Avérous, L. Biobased polyurethanes for biomedical applications. Bioact. Mater. 2021, 6, 1083–1106. [Google Scholar] [CrossRef] [PubMed]
- Debuissy, T.; Pollet, E.; Avérous, L. Biotic and abiotic synthesis of renewable aliphatic polyesters from short building blocks obtained from biotechnology. ChemSusChem 2018, 11, 3836–3870. [Google Scholar] [CrossRef]
- Furtwengler, P.; Avérous, L. Renewable polyols for advanced polyurethane foams from diverse biomass resources. Polym. Chem. 2018, 9, 4258–4287. [Google Scholar] [CrossRef]
- Peyrton, J.; Chambaretaud, C.; Sarbu, A.; Avérous, L. Biobased polyurethane foams based on new polyol architectures from microalgae oil. ACS Sustain. Chem. Eng. 2020, 8, 12187–12196. [Google Scholar] [CrossRef]
- Gholami, H.; Yeganeh, H. Vegetable oil-based polyurethanes as antimicrobial wound dressings: In vitro and in vivo evaluation. Biomed. Mater. 2020, 15, 045001. [Google Scholar] [CrossRef]
- Miao, S.; Wang, P.; Su, Z.; Zhang, S. Vegetable-oil-based polymers as future polymeric biomaterials. Acta Biomater. 2014, 10, 1692–1704. [Google Scholar] [CrossRef] [PubMed]
- Laurichesse, S.; Avérous, L. Chemical modification of lignins: Towards biobased polymers. Prog. Polym. Sci. 2014, 39, 1266–1290. [Google Scholar] [CrossRef]
- Solanki, A.; Das, M.; Thakore, S. A review on carbohydrate embedded polyurethanes: An emerging area in the scope of biomedical applications. Carbohydr. Polym. 2018, 181, 1003–1016. [Google Scholar] [PubMed]
- Usman, A.; Zia, K.M.; Zuber, M.; Tabasum, S.; Rehman, S.; Zia, F. Chitin and chitosan based polyurethanes: A review of recent advances and prospective biomedical applications. Int. J. Biol. Macromol. 2016, 86, 630–645. [Google Scholar] [CrossRef]
- Debuissy, T.; Pollet, E.; Avérous, L. Synthesis and characterization of block poly(ester-ether-urethane)s from bacterial poly(3-hydroxybutyrate) oligomers. J. Polym. Sci. Part. A Polym. Chem. 2017, 55, 1949–1961. [Google Scholar] [CrossRef]
- Oyervides-Munoz, E.; Pollet, E.; Ulrich, G.; de Jesus Sosa-Santillan, G.; Averous, L. Original method for synthesis of chitosan-based antimicrobial agent by quaternary ammonium grafting. Carbohydr. Polym. 2017, 157, 1922–1932. [Google Scholar] [CrossRef] [PubMed]
- Jain-Beuguel, C.; Li, X.; Houel-Renault, L.; Modjinou, T.; Simon-Colin, C.; Gref, R.; Renard, E.; Langlois, V. Water-soluble Poly(3-hydroxyalkanoate) sulfonate: Versatile biomaterials used as coatings for highly porous Nano-metal organic framework. Biomacromolecules 2019, 20, 3324–3332. [Google Scholar] [CrossRef] [PubMed]
- Baheiraei, N.; Gharibi, R.; Yeganeh, H.; Miragoli, M.; Salvarani, N.; Di Pasquale, E.; Condorelli, G. Electroactive polyurethane/siloxane derived from castor oil as a versatile cardiac patch, part I: Synthesis, characterization, and myoblast proliferation and differentiation. J. Biomed. Mater. Res. A 2016, 104, 775–787. [Google Scholar] [CrossRef]
- Buzarovska, A.; Dinescu, S.; Lazar, A.D.; Serban, M.; Pircalabioru, G.G.; Costache, M.; Gualandi, C.; Averous, L. Nanocomposite foams based on flexible biobased thermoplastic polyurethane and ZnO nanoparticles as potential wound dressing materials. Mater. Sci. Eng. C Mater. Biol. Appl. 2019, 104, 109893. [Google Scholar] [CrossRef]
- Dodangeh, F.; Seyed Dorraji, M.S.; Rasoulifard, M.H.; Ashjari, H.R. Synthesis and characterization of alkoxy silane modified polyurethane wood adhesive based on epoxidized soybean oil polyester polyol. Compos. Part. B Eng. 2020, 187, 107857. [Google Scholar] [CrossRef]
- Yuan, Y.; Zhang, Y.; Fu, X.; Jiang, L.; Liu, Z.; Hu, K.; Wu, B.; Lei, J.; Zhou, C. Silane-terminated polyurethane applied to a moisture-curable pressure-sensitive adhesive using triethoxysilane. RSC Adv. 2016, 6, 83688–83696. [Google Scholar]
- Heck, C.A.; Giacomolli, D.A.; Livotto, P.R.; dos Santos, J.H.Z.; Wolf, C.R. Hybrid silica generatedIn situin polyurethane-based composites. J. Appl. Polym. Sci. 2014, 131. [Google Scholar] [CrossRef]
- Radev, L.; Zheleva, D.; Michailova, I. In vitro bioactivity of Polyurethane/85S Bioglass composite scaffolds. Open Chem. 2013, 11. [Google Scholar]
- Rashti, A.; Yahyaei, H.; Firoozi, S.; Ramezani, S.; Rahiminejad, A.; Karimi, R.; Farzaneh, K.; Mohseni, M.; Ghanbari, H. Development of novel biocompatible hybrid nanocomposites based on polyurethane-silica prepared by sol gel process. Mater. Sci. Eng. C Mater. Biol. Appl. 2016, 69, 1248–1255. [Google Scholar] [CrossRef]
- Gao, F.; Bin, K.; Huang, S.-Q. Synthesis and characterization of bis-triethoxysilane endcapped polyurethane/urea. J. Appl. Polym. Sci. 2011, 122, 798–803. [Google Scholar] [CrossRef]
- Rahman, M.M.; Hasneen, A.; Lee, W.-K.; Lim, K.T. Preparation and properties of sol-gel waterborne polyurethane adhesive. J. Sol.-Gel Sci. Technol. 2013, 67, 473–479. [Google Scholar] [CrossRef]
- Duan, Y.; Jana, S.C.; Lama, B.; Espe, M.P. Reinforcement of Silica Aerogels Using Silane-End-Capped Polyurethanes. Langmuir 2013, 29, 6156–6165. [Google Scholar] [CrossRef] [PubMed]
- Levy, D.; Zayat, M. The Sol.-Gel Handbook; John Wiley & Sons: Hoboken, NJ, USA, 2015; pp. 1–1508. [Google Scholar]
- Gharibi, R.; Yeganeh, H.; Rezapour-Lactoee, A.; Hassan, Z.M. Stimulation of wound healing by electroactive, antibacterial, and antioxidant polyurethane/siloxane dressing membranes: In vitro and in vivo evaluations. ACS Appl. Mater. Interfaces 2015, 7, 24296–24311. [Google Scholar] [CrossRef]
- Song, E.-H.; Jeong, S.-H.; Park, J.-U.; Kim, S.; Kim, H.-E.; Song, J. Polyurethane-silica hybrid foams from a one-step foaming reaction, coupled with a sol-gel process, for enhanced wound healing. Mater. Sci. Eng. C 2017, 79, 866–874. [Google Scholar] [CrossRef] [PubMed]
- Gharibi, R.; Yeganeh, H.; Gholami, H.; Hassan, Z.M. Aniline tetramer embedded polyurethane/siloxane membranes and their corresponding nanosilver composites as intelligent wound dressing materials. RSC Adv. 2014, 4, 62046–62060. [Google Scholar] [CrossRef]
- Nayyer, L.; Birchall, M.; Seifalian, A.M.; Jell, G. Design and development of nanocomposite scaffolds for auricular reconstruction. Nanomedicine 2014, 10, 235–246. [Google Scholar] [CrossRef] [PubMed]
- Wendels, S.; Heinrich, B.; Donnio, B.; Avérous, L. Green and controlled synthesis of short diol oligomers from polyhydroxyalkanoate to develop fully biobased thermoplastics. Eur. Polym. J. 2021, 153, 110531. [Google Scholar] [CrossRef]
- Laurichesse, S.; Huillet, C.; Avérous, L. Original polyols based on organosolv lignin and fatty acids: New bio-based building blocks for segmented polyurethane synthesis. Green Chem. 2014, 16, 3958–3970. [Google Scholar] [CrossRef]
- Calvo-Correas, T.; Martin, M.D.; Retegi, A.; Gabilondo, N.; Corcuera, M.A.; Eceiza, A. Synthesis and Characterization of Polyurethanes with High Renewable Carbon Content and Tailored Properties. ACS Sustain. Chem. Eng. 2016, 4, 5684–5692. [Google Scholar] [CrossRef]
- Chenal, J.-M.; Chazeau, L.; Guy, L.; Bomal, Y.; Gauthier, C. Molecular weight between physical entanglements in natural rubber: A critical parameter during strain-induced crystallization. Polymer 2007, 48, 1042–1046. [Google Scholar] [CrossRef]
- Palmese, G.R.; McCullough, R.L. Effect of epoxy-amine stoichiometry on cured resin material properties. J. Appl. Polym. Sci. 1992, 46, 1863–1873. [Google Scholar] [CrossRef]
- Bueno-Ferrer, C.; Hablot, E.; Garrigós, M.d.C.; Bocchini, S.; Averous, L.; Jiménez, A. Relationship between morphology, properties and degradation parameters of novative biobased thermoplastic polyurethanes obtained from dimer fatty acids. Polym. Degrad. Stab. 2012, 97, 1964–1969. [Google Scholar] [CrossRef]
- Javni, I.; Petrović, Z.S.; Guo, A.; Fuller, R. Thermal stability of polyurethanes based on vegetable oils. J. Appl. Polym. Sci. 2000, 77, 1723–1734. [Google Scholar] [CrossRef]
- Mikhailova, A.; Tamboura, M.; Jia, M. Synthesis, characterization, and analyses of mechanical, adhesion, and thermal properties of polysiloxane resin modified with segmented polyurethane. J. Coat. Technol. Res. 2012, 10, 97–108. [Google Scholar] [CrossRef]
- Hamdani-Devarennes, S.; Longuet, C.; Perrin, D.; Lopez-cuesta, J.-M.; Ganachaud, F. Flame retardancy of silicone-based materials. Polym. Degrad. Stab. 2009, 94, 465–495. [Google Scholar] [CrossRef]
- Monge, S.; Zhang, X.; Giani, O.; Robin, J.-J. Organic/inorganic hybrid materials from polypeptide-based block copolymers. React. Funct. Polym. 2009, 69, 380–384. [Google Scholar] [CrossRef]
- Hagen, R.; Salmén, L.; Lavebratt, H.; Stenberg, B. Comparison of dynamic mechanical measurements and Tg determinations with two different instruments. Polym. Test. 1994, 13, 113–128. [Google Scholar] [CrossRef]
- Fu, C.; Yang, Z.; Zheng, Z.; Shen, L. Properties of alkoxysilane castor oil synthesized via thiol-ene and its polyurethane/siloxane hybrid coating films. Prog. Org. Coat. 2014, 77, 1241–1248. [Google Scholar] [CrossRef]
- Batich, C.; DePalma, D.; Marotta, J.; Latorre, G. Silicone Degradation Reactions; Immunology of Silicones, Berlin, Heidelberg, 1996; Potter, M., Rose, N.R., Eds.; Springer: Berlin/Heidelberg, Germany, 1996; pp. 13–23. [Google Scholar]
- Barrioni, B.R.; de Carvalho, S.M.; de Oliveira, A.A.R.; Pereira, M.d.M. Improved biocompatibility of polyurethane film by association with bioactive glass through ultrasonic implantation. Mater. Lett. 2018, 223, 53–56. [Google Scholar] [CrossRef]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Xerogel | Series N° | Di-isocyanate | PU | SPU | Xerogels | Biobased Content [wt%] | |||
|---|---|---|---|---|---|---|---|---|---|
| Name | NCO/OH Ratio | Name | APTMS Molar Eq. | Rw | TEOS/SPU Ratio | ||||
| XP2-1R1 | 2 | HDI | PUP2-1 | 2 | SPU2-1 | 1 | 1 | 0 | 26 |
| XP2-1R2 | 1, 2, 3 and 4 | HDI | PUP2-1 | 2 | SPU2-1 | 1 | 2 | 0 | 26 |
| XP2-1R8 | 2 | HDI | PUP2-1 | 2 | SPU2-1 | 1 | 8 | 0 | 26 |
| XP4-1R2 | 1 | HDI | PUP4-1 | 4 | SPU4-1 | 3 | 2 | 0 | 12 |
| XP6-1R2 | 1 | HDI | PUP6-1 | 6 | SPU6-1 | 5 | 2 | 0 | 8 |
| XPD2-1R2 | 3 | DDI | PUPD2-1 | 2 | SPUD2-1 | 1 | 2 | 0 | 78 |
| XPT3-1 | 4 | HDI | PUP2-1 | 2 | SPU2-1 | 1 | 2 | 0.33 | 17 |
| XPT1-1 | 4 | HDI | PUP2-1 | 2 | SPU2-1 | 1 | 2 | 1.00 | 13 |
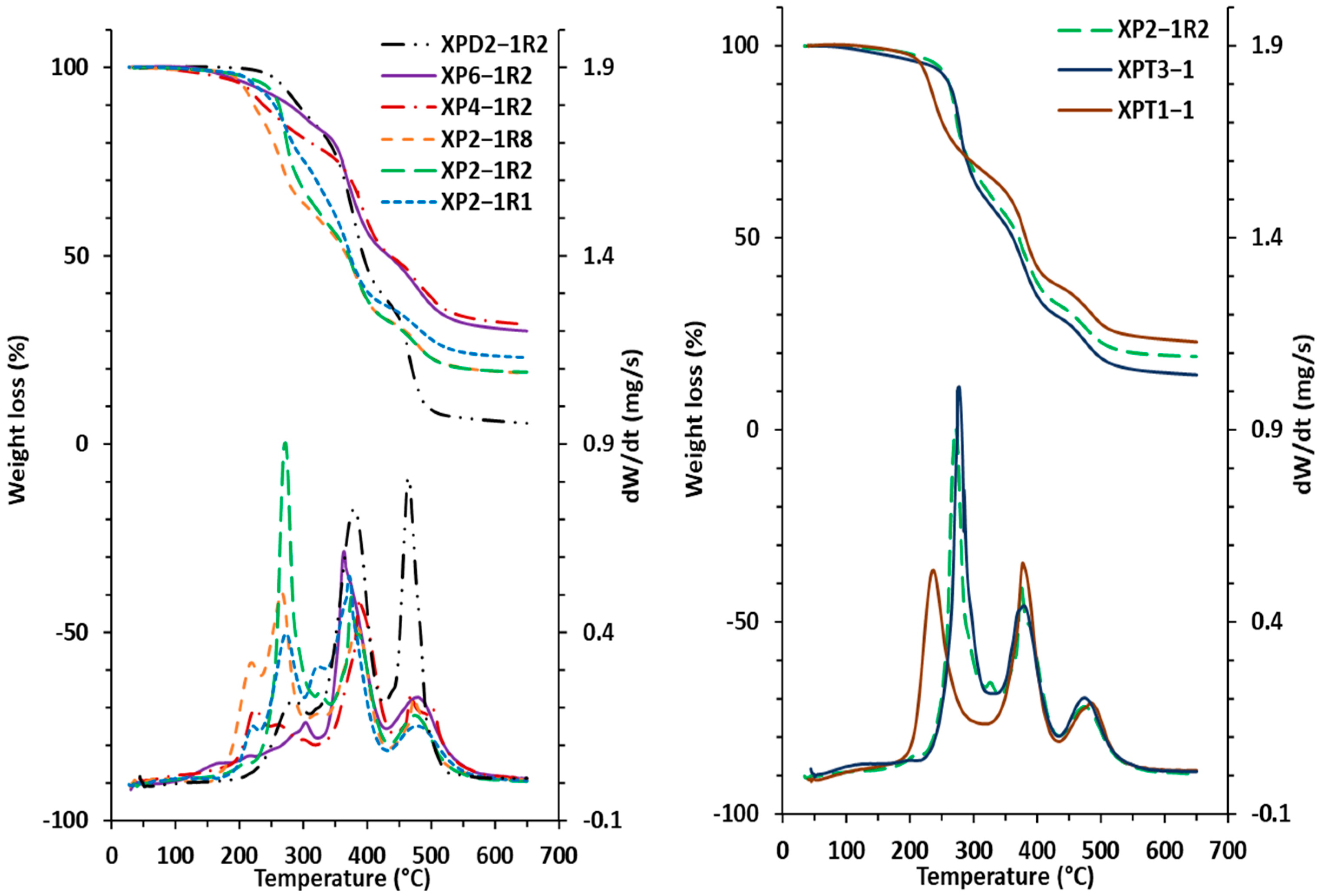
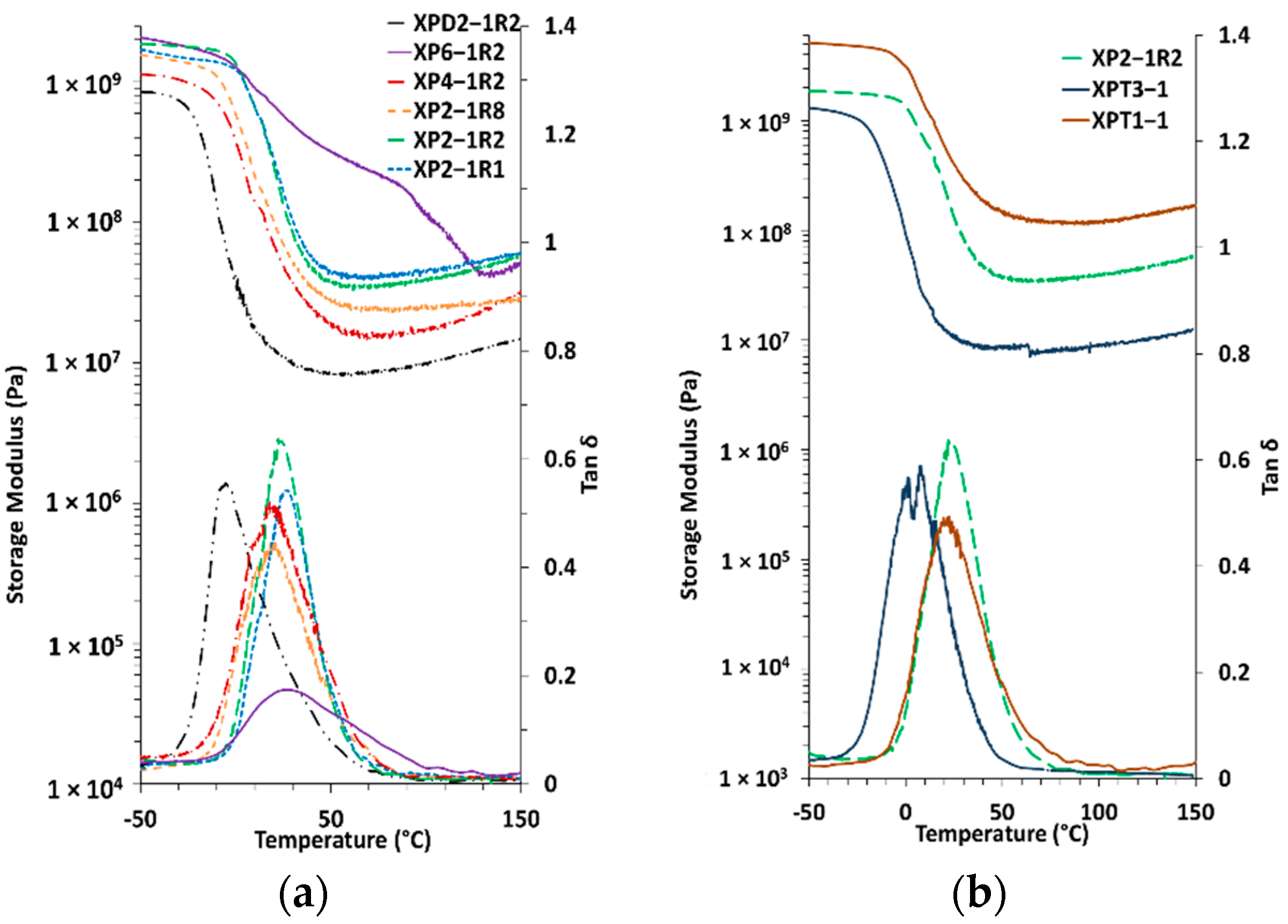
| Xerogel | TGA T5% [ °C] | TGA Td1,max [ °C] | TGA Td2,max [ °C] | TGA Td3,max [ °C] | Char Residue at 650 °C [%] | DSC Tg [ °C] | DMA Tα [ °C] | νe [mol.m−3] | Mc [g.mol−1] |
|---|---|---|---|---|---|---|---|---|---|
| XP2-1R1 | 227 | 271 | 372 | 478 | 22.9 | 15 | 27 | 4900 | 301 |
| XP2-1R2 | 242 | 271 | 375 | 476 | 19.1 | 18 | 22 | 4269 | 349 |
| XP2-1R8 | 206 | 266 | 385 | 473 | 18.7 | 9 | 21 | 2680 | 612 |
| XP4-1R2 | 209 | 261 | 389 | 482 | 31.8 | 11 | 18 | 1910 | 670 |
| XP6-1R2 | 222 | 304 | 363 | 486 | 29.6 | 20 | 28 | 8508 | 170 |
| XPD2-1R2 | 268 | 286 | 380 | 465 | 5.5 | −14 | −5 | 986 | 1617 |
| XPT3-1 | 227 | 276 | 378 | 475 | 14.3 | 17 | 4 | 926 | 1382 |
| XPT1-1 | 217 | 236 | 377 | 485 | 22.9 | 16 | 23 | 13234 | 118 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wendels, S.; de Souza Porto, D.; Avérous, L. Synthesis of Biobased and Hybrid Polyurethane Xerogels from Bacterial Polyester for Potential Biomedical Applications. Polymers 2021, 13, 4256. https://doi.org/10.3390/polym13234256
Wendels S, de Souza Porto D, Avérous L. Synthesis of Biobased and Hybrid Polyurethane Xerogels from Bacterial Polyester for Potential Biomedical Applications. Polymers. 2021; 13(23):4256. https://doi.org/10.3390/polym13234256
Chicago/Turabian StyleWendels, Sophie, Deyvid de Souza Porto, and Luc Avérous. 2021. "Synthesis of Biobased and Hybrid Polyurethane Xerogels from Bacterial Polyester for Potential Biomedical Applications" Polymers 13, no. 23: 4256. https://doi.org/10.3390/polym13234256