1. Introduction
Polyurethane foam composites are one of the most popular polymeric materials. They are polymers prepared by the reaction of polyether polyols or polyester polyols with binary isocyanate or polyisocyanates, with carbamate (-NH-COO-) repeating structural units. They have a wide range of applications in construction, refrigeration, and insulation, among others, because of their good electrical conductivity, low thermal conductivity, mechanical properties, and good shock absorption.
Polyester polyols and polyether polyols are important chemical intermediates in the production of polyurethane materials. However, at present, the raw materials of polyester and polyether polyols consist of toxic and corrosive petrochemical products, such as phthalic anhydride and phthalic acid. The use of such non-renewable oil resources has resulted in pollution and threats to the natural environment. The most effective way to address this issue is to replace polyester polyols or polyether polyols with renewable woody biological raw materials to prepare green, environmentally friendly and biodegradable biomass-based materials, such as biomass-based polyurethane foam [
1].
Lignocellulose includes lignin, cellulose and hemicellulose as well as a small amount of organic matter such as fat, protein, pectin and ash. Lignocellulosic biomass is not only a renewable, abundant and cheap resource with a large amount of hydroxyl groups, but also has a stable three-dimensional network structure, which can replace and modify petroleum polyols in chemical production. Lima García et al. [
2] found that high purity lignin dispersed to sufficiently small particle size can improve the lap shear strength and the wood failure percentage of bonded beech specimens as well as the gap filling properties of resin. Manggar et al. [
3] successfully prepared high strength, low curing temperature, short pressing time, and isocyanate-free lignin-based non-isocyanate polyurethane resins. Tavares et al. [
4] prepared polyurethanes based on renewable raw materials (technical Kraft lignin, castor oil and modified castor oil), and the results indicated that the starting materials change the polymer’s properties. The oil modification process improved hydroxyl concentration and led to polyurethanes with enhanced mechanical properties versus ones synthesized from unmodified castor oil. The products show the feasibility of developing polyurethane-type materials with large property ranges, by using an industrial, low-cost, unmodified and largely available residue combined with a non-edible renewable source oil. Ravindra et al. [
5] successfully used lignins as part of the polyol to synthesize lignin-polyurethane-based wood adhesives. A slower setting time and better adhesion to the wood substrate was observed as compared to the standard PU, and the toxicity was also reduced. These results support the development of polyurethane adhesive using lignin as a natural and renewable polyol, allowing the reuse of this industrial waste. Lignocellulose fiber has low density, high specific strength and stiffness, good heat insulation and sound insulation performance; it is widely used in the toughening modification of various materials. Poplar is a common fast-growing tree species in China because of its light weight and fast growth rate. Poplar fiber has been widely used in composite materials because of its low cost, high yield, easy processing, high elasticity, long fibers and high content. Therefore, in this study, wood fibers of white poplar were used to toughen polyurethane foams [
6]. Lignocellulose, as a green, natural material, not only can reduce the use of petroleum polyols, but also can give polyurethane materials biodegradable performance, reduce the secondary pollution of products, and bring new hope for the development of the polyurethane industry.
At present, the application of lignocellulose in polyurethane materials includes two main aspects: on the one hand, lignocellulose is directly used as a compatibilizer or filler material; on the other hand, lignocellulose is chemically modified or liquefied to produce polyols as the raw material for polyurethane preparation [
7]. The composition of lignocellulose is complex, and the structure and physicochemical properties of its three-dimensional network polymers are different due to the different sources and separation methods. Therefore, it is difficult to directly prepare polyurethane materials using lignocellulose as a compatibilizer or filler [
8]. Compared with chemical modification, liquefaction is simpler and also one of the most effective methods for resource utilization. Liquefaction under atmospheric pressure, in particular, has attracted much attention because of its mild reaction conditions and remarkable liquefaction effect [
9]. Liquefaction technology can convert the chemical composition of biomass into a liquid polymer material, which can be used in the production of adhesives and foams [
10]. It is a thermochemical process of converting wood biomaterials directly from solid state to liquid state with the help of a liquefied solvent under certain temperature and pressure conditions.
By means of liquefaction, natural polymers such as cellulose, hemicellulose and lignin in lignocellulose can be degraded into liquid products with relatively low molecular weight and certain reactivity. These liquefied products can be further used as fuel or chemical raw materials [
11]. The liquefaction of woody biomass under atmospheric pressure can be realized by selecting proper solvents as liquefaction reagents. Compared with high-temperature and high-pressure liquefaction methods, liquefaction under atmospheric pressure is a mild liquefaction method [
8].
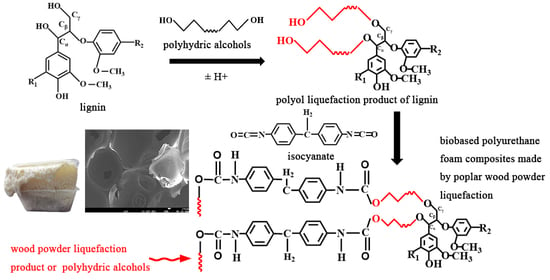
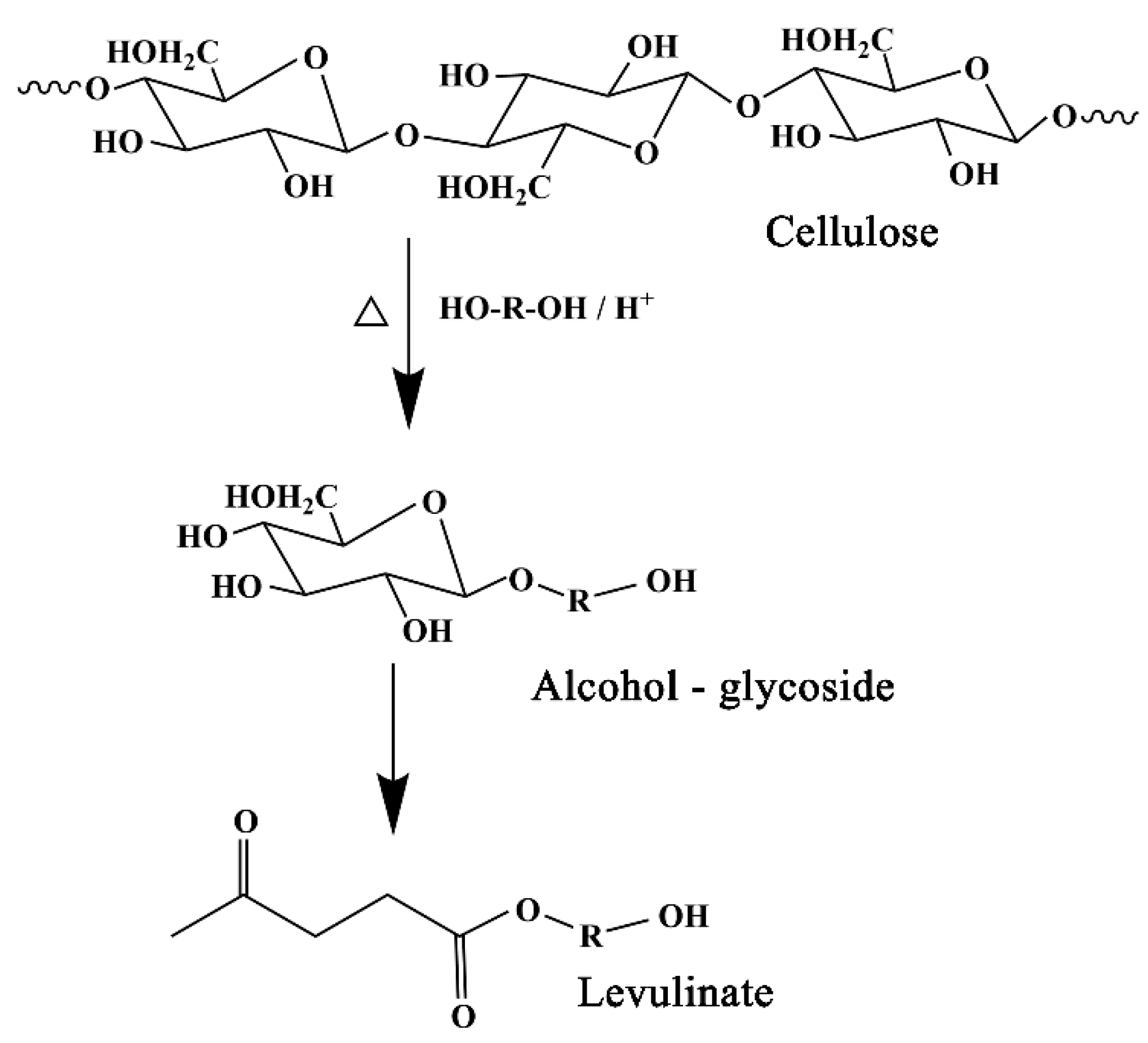
In practical application, different liquefaction solvents are often chosen according to the final use of liquefied products. If polyhydroxy alcohols, such as ethylene glycol (EG), polyethylene glycol (PEG) or a mixture of the two solvents, are used as liquefaction solvents, the liquefaction products are mainly used to prepare polyurethane adhesives or polyurethane foams. If phenol is used as the liquefaction solvent, its liquefaction products are usually used to prepare thermoplastic and thermosetting phenolic resins, foaming materials, molded materials, carbon fibers, etc. [
12].
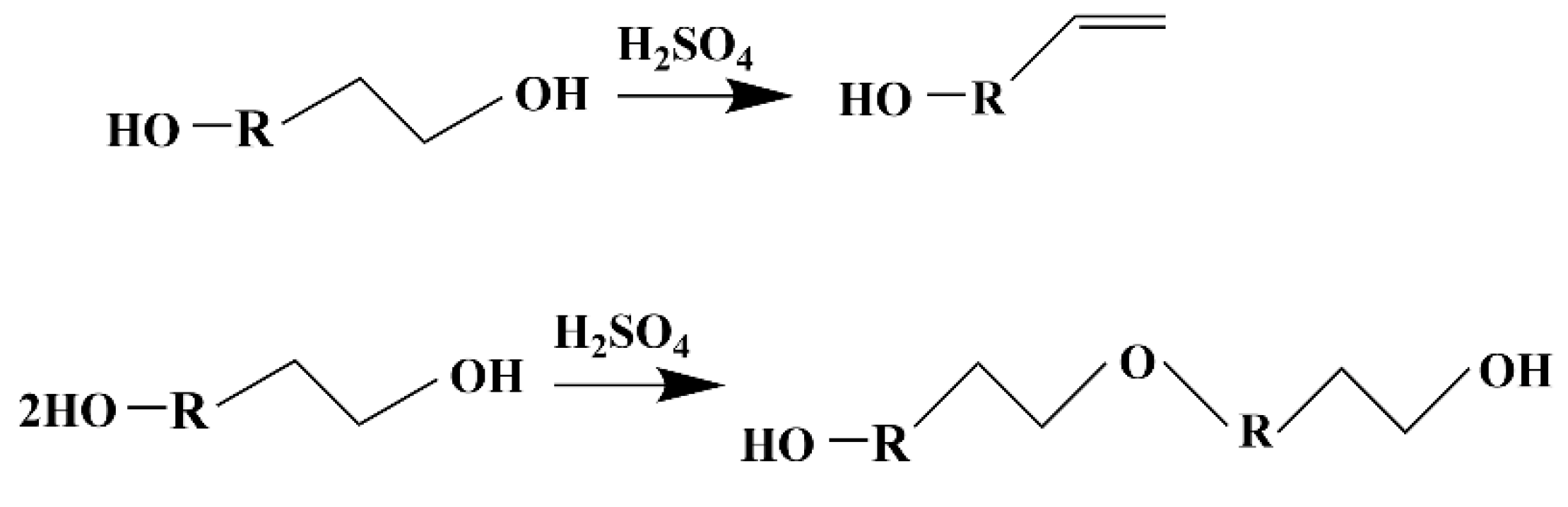
Polyethylene glycol (PEG) is usually used as the liquefaction reagent in the liquefaction of lignocellulosic biomass. Because polyethylene glycol itself can be polymerized with an isocyanate monomer, the liquefied products of lignin biomass can be directly used in the preparation of polyurethane materials without any separation. When polyethylene glycol is used as a liquefaction reagent, it is usually necessary to add a small amount of small molecule polyhydroxyl compounds (ethylene glycol, glycerol, etc.) as an auxiliary liquefaction reagent to inhibit the condensation reaction [
8]. Kurimoto et al. found that the addition of 11.1% glycerol to polyethylene glycol could reduce the residue rate and delay the occurrence of polycondensation reaction, the addition of 33.3% glycerol could inhibit the occurrence of polycondensation reaction, and the addition of glycerol improved the availability of wood liquefaction products [
13].
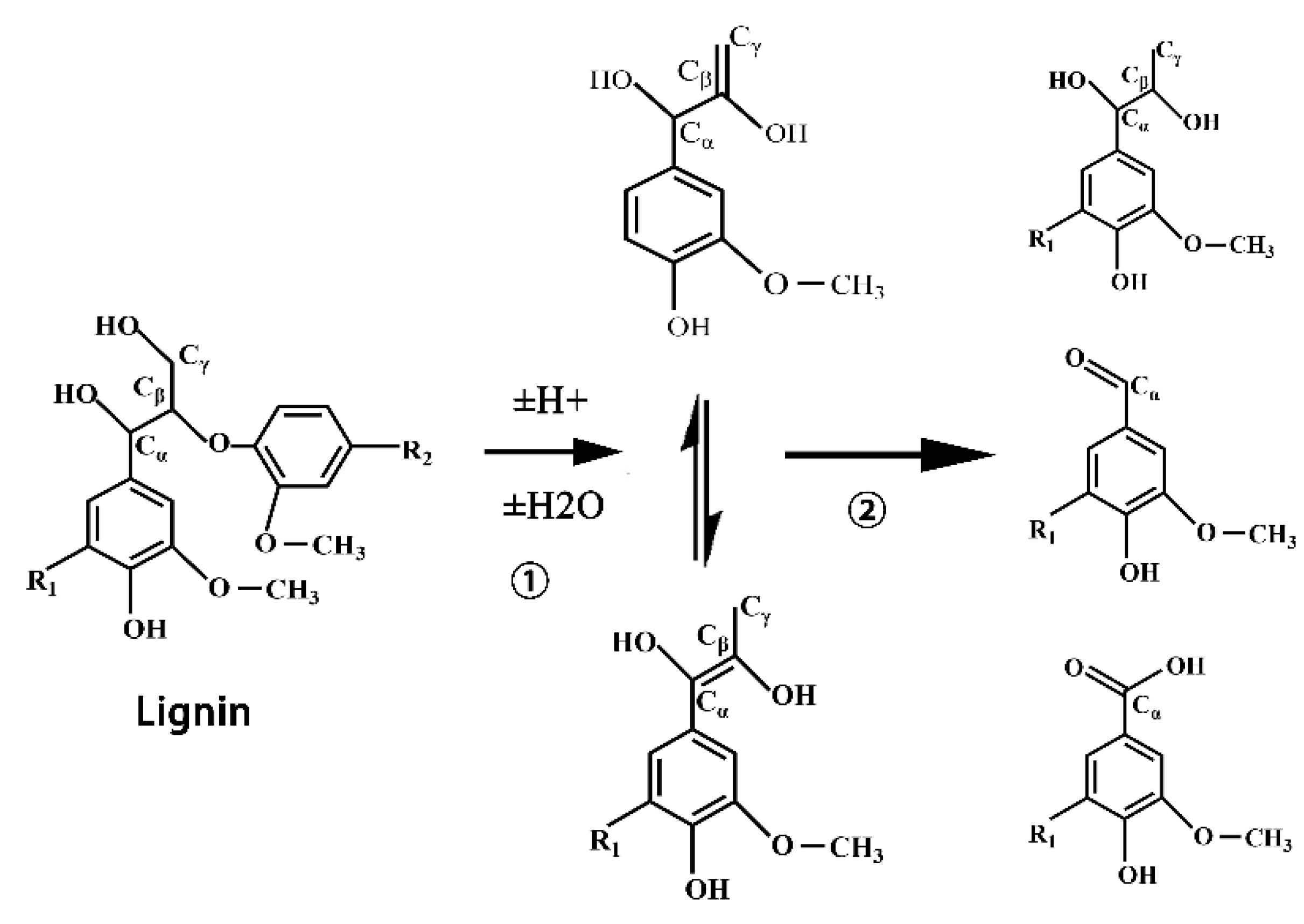
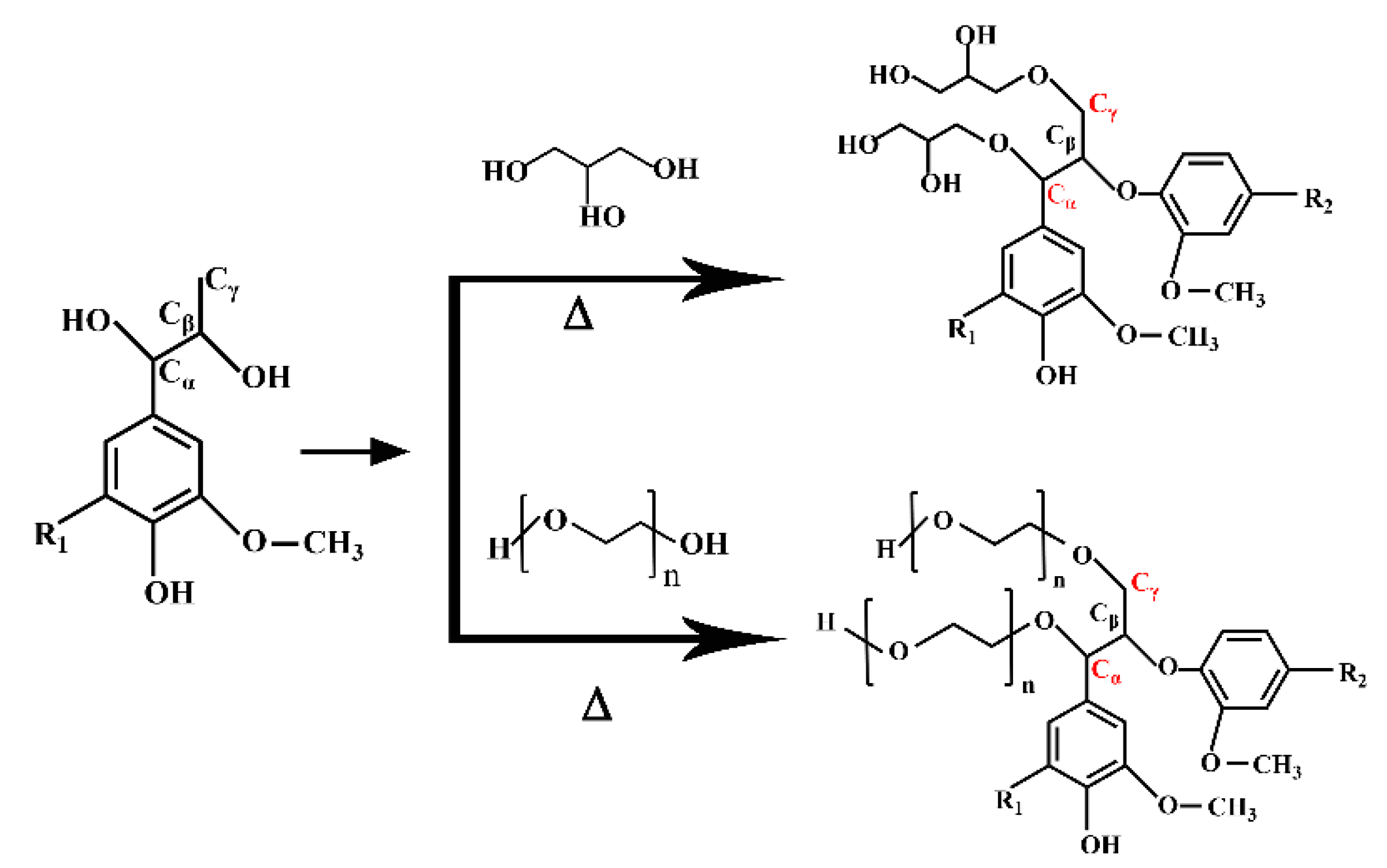
When powdered lignocellulosic macromolecules are liquefied to obtain reactive small molecules, which are then added to the preparation of polyurethane foam, the compatibility between liquefied products and isocyanates is increased, and the properties of polyurethane are also improved. However, due to the structure and morphology of different biomasses, a large number of complex chemical reactions take place simultaneously and compete with each other in the liquefaction process of wood powder [
14]. Shiraishi et al. [
15] used hydroxy-propylated wood solution to prepare polyurethane foam. The results showed that although the foam had high strength and good compressive deformation recovery ability, its apparent density was as low as 0.04 g/cm
3 and thus its mechanical properties were insufficient. Consequently, the liquefaction condition of wood powder is an important element. The liquefaction of wood with polyether polyols or polyester polyols may further improve the properties of the foamed plastics. As a result, it is of great practical significance to study the complex chemical changes in different stages of wood powder liquefaction and find the optimal liquefaction conditions for the preparation of wood-powder-based polyurethane foam, which can replace petroleum polyols with biomass energy, optimize polyurethane properties and reduce costs.
In this paper, the liquefaction mechanism of different lignocellulose components under different liquefaction times as well as the effects of different liquefaction times on the structure, surface morphology, apparent density and compressive strength of polyurethane foam were studied.
2. Materials and Methods
NaOH (98%), glycerol, polyethylene glycol 400 (PEG400, average molecular weight of 400), concentrated sulfuric acid (mass fraction 98%), phthalic anhydride and pyridine were provided by Lan-yi Chemical Products Co. Ltd., Beijing, China.
The wood powder of white poplar (40 mesh) was provided by Chengzun Mineral Products Processing limited company, Ling-shou County, Shijiazhuang, China.
The following were provided by Bailing Qingyue Polyurethane Company, Beijing, China: diphenylmethylene diisocyanate (MDI), industrial grade, isocyanate content of 30.31%; polyether polyols 4110 and polyether polyols 403, industrial grade; triethylenediamine solution (A33), industrial grade; dimethyl methyl phosphate (DMMP), industrial grade; silicone oil, industrial grade; 1, 1-Dichloro-1-fluoroethane (HCFC-141B), industrial grade.
2.1. Preparation of Poplar-Powder-Based Polyols
2.1.1. Instruments and Equipment
The collection type constant temperature heating magnetic agitator DF-101S, Bangxi Instrument Technology (Shanghai, China) Co., LTD, was used to stir the sample solution evenly.
A multi-function electric stirrer, RCT B S025, IKA, Germany, was used to stir the wood powder and liquefy the solvent when liquefying the wood powder.
A vacuum drying oven, DZF-6053, Yi-heng Scientific Instrument Co., Ltd., Shanghai, China, was used to mature the polyurethane foam.
2.1.2. Methods
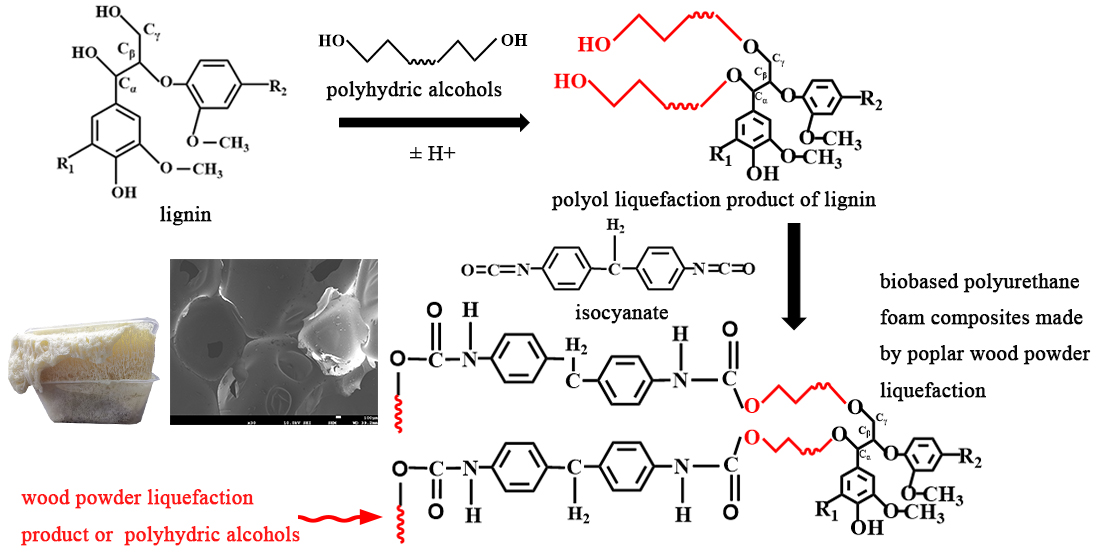
A solvent mixture of poplar wood powder, PEG400 and glycerol and catalyst sulfuric acid were added in proportion to a separable flask containing an agitator, thermometer and reflux condenser. Then, the separable flask was dipped into a pre-heated oil bath and stirred for a period of time. After the reaction, the separation flask was immediately placed in a beaker with ice water to cool down and stop the reaction. NaOH was added to the flask and the value of pH of the solution was adjusted to 7 and let stand overnight. The residue was filtered out, and the supernatant was poplar-powder-based polyols.
This experiment used PEG/glycerol 4:1 (W/W), liquid-solid ratio 9:1, liquefaction temperature 130 °C, sulfuric acid (catalyst) dosage 1.8% (sulfuric acid in the mass ratio of liquefaction solvent and wood powder raw material), liquefied for 20 min, 50 min, 80 min, 110 min, 140 min.
2.1.3. Determination of Hydroxyl Value of Liquefied Products
The hydroxyl value of the liquefied product was determined with reference to GB/T12008.3-2009 [
16], “Plastic polyether polyols—Part 3: Determination of hydroxyl value”. The principle method used was as follows: the hydroxyl in the sample is esterified by reflux in pyridine solution of phthalic anhydride; imidazole is used as the catalyst; the excessive anhydride is hydrolyzed in water; the phthalic acid generated is titrated with sodium hydroxide standard titration solution; and the hydroxyl value is calculated by the difference between the sample and blank titration.
The phthalic anhydride acylation reagent was prepared as follows: weave 116 g of phthalic anhydride and dissolve in a 1 L brown bottle; add 700 mL of pyridine and vigorously shake until dissolved; add 16 g of imidazole and carefully shake until dissolved. The solution should be left to rest overnight before use.
The methods used were as follows: Sample 1 to 1.5 g with a syringe into a conical flask, and add 25 mL of phthalic anhydride acylating agent to each sample and blank conical flask using a pipette. Shake the flask until the test material is dissolved. Connect each conical flask with an air-condensing tube and place in an oil bath at (115 ± 2) °C for reflux for 30 min; shake the flask 1~2 times during reflux. After heating, remove the device from the oil bath and cool to room temperature. Rinse the condensing tube drop by drop with 30 mL pyridine and remove the condensing tube. Quantitatively transfer the solution to a 250 mL beaker and rinse the flask with 20 mL pyridine. Stir the beaker on a magnetic stirrer and determine the hydroxyl value of the liquefied product by potentiometric titration. Titrate the solution with a standard titration solution of 0.5 mol/L NaOH to pH = 7. Conduct the blank test in the same way. Calculate the hydroxyl value (
OHV) according to Equation (1):
V3: the volume of the NaOH standard titration solution consumed when titrating the sample, mL;
V4: the volume of the NaOH standard titration solution consumed when titrating the blank, mL;
c: the concentration of the NaOH standard titration solution, mol/L;
m: quality of test material, g;
56.1: molar mass of KOH, g/mol.
2.1.4. Determination of Acid Number of Liquefied Products
The acid number of the liquefied product was determined by referring to GB/T12008.5-2010 [
17], “Plastics polyether polyols—Part 5: Method for determination of acid value”. The principle method used was as follows: dissolve the sample in isopropyl alcohol, make phenolphthalein an indicator, and add 0.02 mol/L potassium hydroxide methanol standard solution to the terminal point at room temperature.
The conical flask was filled with (100 ± 20) mL isopropyl alcohol and 1 mL phenolphthalein indicator solution. The solution was titrated to light pink with 0.02 mol/L potassium hydroxide methanol standard solution for 30 s. A total of 50~60 g of test material was deposited into the conical flask and the quality of the test material was recorded, accurate to 0.1 g. The solution in the conical flask was shaken until the sample was completely dissolved. The sample solution was titrated with 0.02 mol/L potassium hydroxide-methanol standard solution. The end point was determined by potentiometric titration, and the volume consumed was recorded.
The acid value C of the sample was measured by the amount of KOH consumed, in mg/g, and calculated according to Equation (2):
A—volume of potassium hydroxide-methanol standard titration solution consumed by titration test material, mL;
N—concentration of potassium hydroxide-methanol standard titration solution, mol/L;
56.1—molar mass of KOH, g/mol;
W—quality of test material, g.
2.1.5. Determination of Relative Molecular Weight and Distribution of Liquefied Products
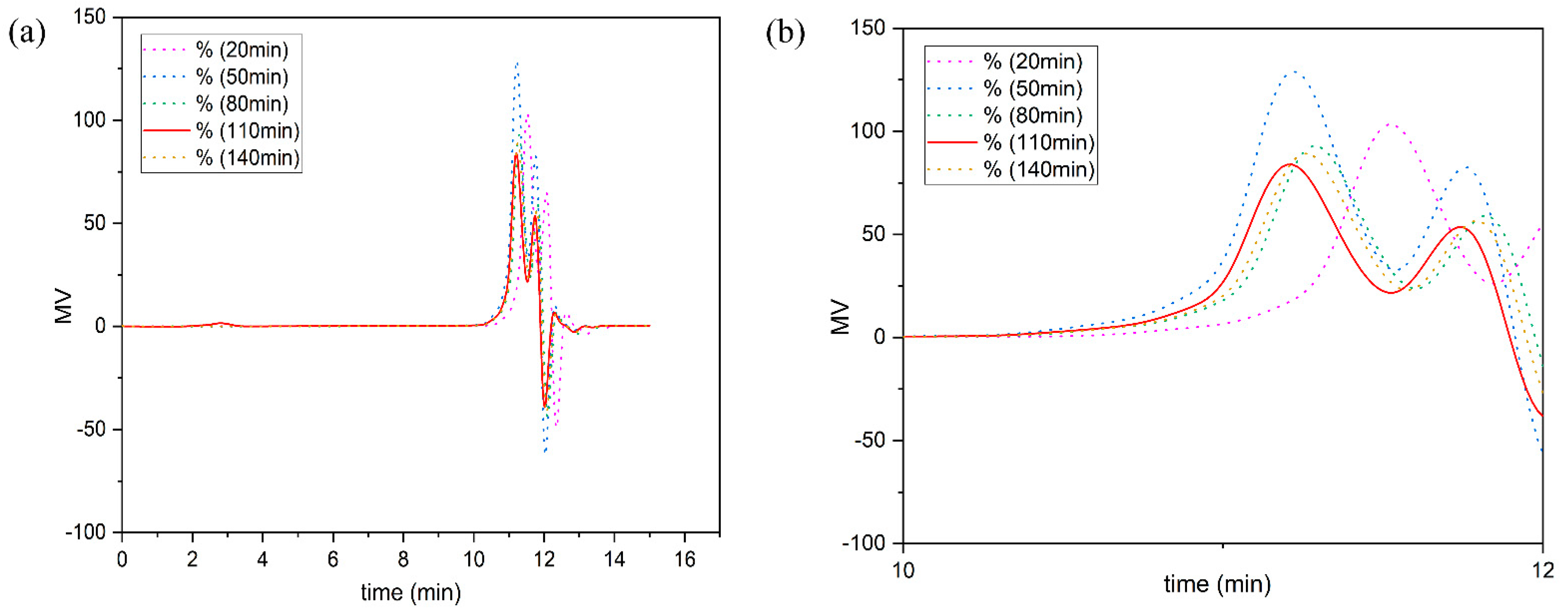
The relative molecular weight and distribution of the liquefied products removed from the residue were determined using a 1525 high-performance liquid chromatograph, Waters Company, US, and PL-GPC220 high-temperature gel penetration chromatograph, Agilent Company, US. The analysis conditions were as follows: the mobile phase was tetrahydrofuran, the flow rate was 1 mL/min, the detector temperature was 30 °C, the standard sample was polystyrene and the molecular weight range was 500–3,000,000.
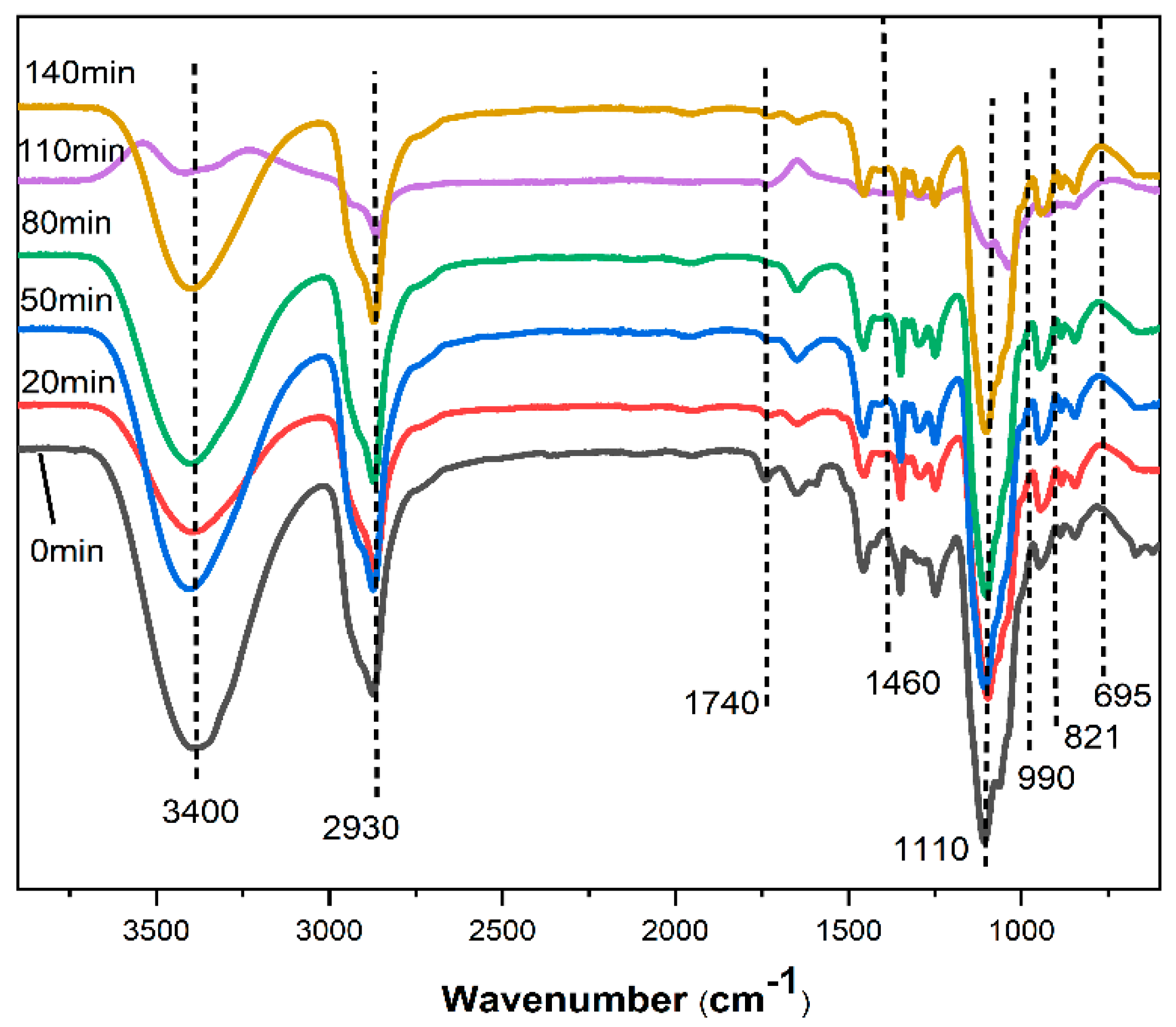
2.1.6. Fourier Transform Infrared Spectroscopy (FT-IR) Spectroscopic Evaluations of Liquefied Products
The Nicolet 6700 Fourier transform infrared spectrometer from American Nicolet Instrument Company was used. The determination resolution was 4 cm−1, the number of scans was 64 and the spectral range was 400–4000 cm−1.
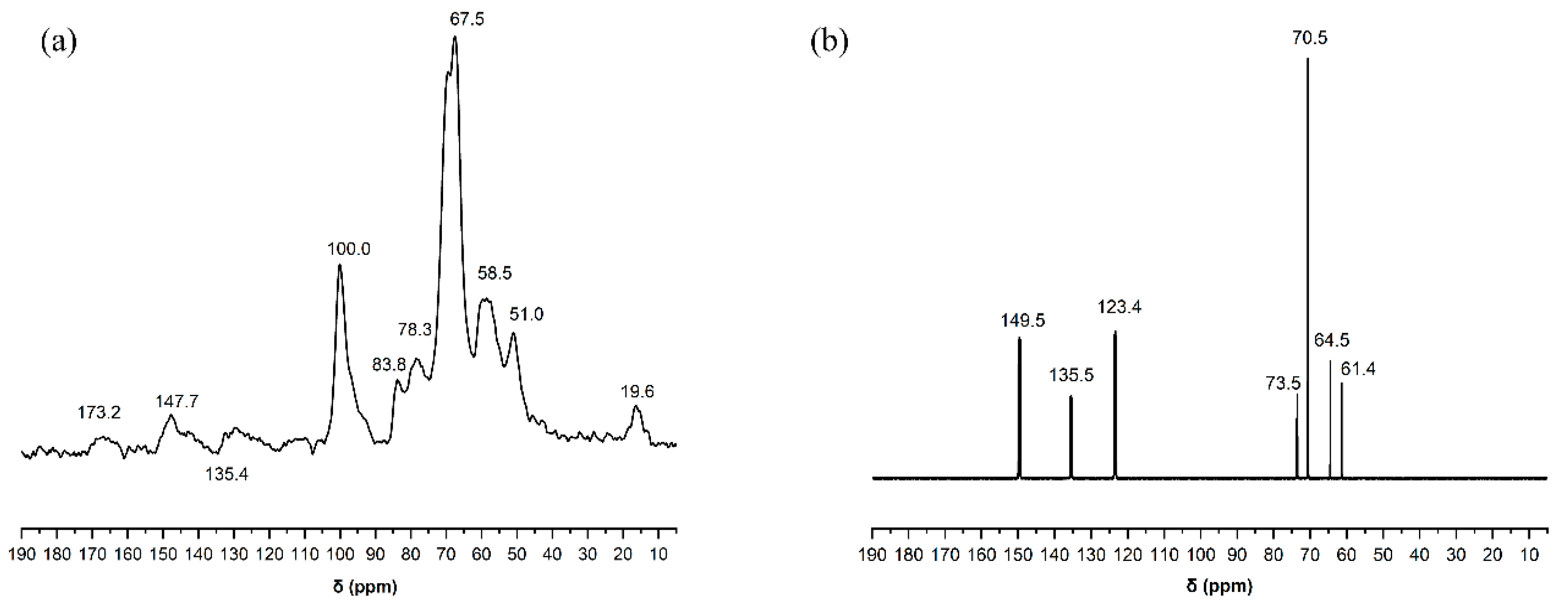
2.1.7. Nuclear Magnetic Resonance (NMR) Spectroscopy of Liquefied Products
The ASCEND 400 liquid NMR instrument, Bruker, Germany, was used under the following 13C-NMR measurement conditions. The solvent was pyridine, the frequency was 150.92 MHz, the number of scanning times was 6144, the pulse angle was 90°, the pulse width was 4.50 μs, the capture time was 0.56 s and the delay time was 2 s. Total test time was 13 h.
2.2. Preparation and Characterization of Polyurethane Foam Composites Based on Liquefied Poplar Wood Powder
2.2.1. Material Calculation in the Preparation Process of Liquefied Modified Poplar-Based Polyurethane Foaming Material
The chain expansion, foaming, crosslinking and other reactions occurring in the preparation of polyurethane foaming materials all involve the material ratio, and the correct calculation of material ratio is key to ensure the synthesis of polyurethane foaming materials. If the amount of isocyanate is too small, that is, it is lower than the theoretical calculation value, it will cause insufficient growth of the polymer chain or cause an insufficient amount of CO
2 produced by reaction with the water, which will directly affect the mechanical properties and density of the foam. If the amount of isocyanate is too large, it will not only lead to the wasting of raw materials but also lead to a fast reaction rate and fast chain growth rate, resulting in the formation of too many crosslinking bonds, such as biuret and urea-formate, which will seriously affect the balance between the foaming rate and gel rate, resulting in the phenomenon of brittle and hard foams [
18].
The type and dosage of each component in the formula were determined according to the literature as well as development and production experience. As shown in
Table 1.
After determining the dosage of polyols, catalysts, foaming agents and foaming stabilizers, the dosage of isocyanate components was calculated. The amount of isocyanate used in this study refers to the total amount of isocyanate required in the one-step synthesis process of polyols and water. The amount of isocyanate is calculated using Equation (3).
MMDI: number of substances containing the isocyanate group per gram of MDI, mmol/g;
WMDI: mass of isocyanate MDI, g;
MLL: amount of hydroxyl in the liquefied product per gram, mmol/g;
WLL: mass of the liquefied product, g;
Mpolyol: amount of hydroxyl in polyether polyols per gram, mmol/g;
Wpolyol: quality of polyether polyols, g;
Wwater: quality of the water, g.
The total amount of isocyanate used in the foaming formula, in addition to the amount required in the above formula, should also consider the degree and purity of the excess isocyanate used in the foaming process. Therefore, the amount of isocyanate used in the foaming formulation is generally greater than that required in Equation (3).
2.2.2. Synthesis Formula and Preparation Method of Liquefied Modified Poplar-Based Polyurethane Foaming Material
Polyether polyols 4110 (40 g) and 403 (60 g) were added into the paper cup at a certain ratio, and a quantitative amount of poplar-based polyols was added. The polyols were dried in a vacuum drying oven at 70 °C for 10 min. The catalyst triethylene diamine (A33) and dibutyltin dilaurate, foam stabilizer silicone oil, flame retardant dimethyl methyl phosphate (DMMP), foaming agent 1,1-dichloro-1-fluorine ethane (HCF-141B) and a small amount of water were successively added. The isocyanate was added and stirred vigorously for 10~15 s under an electric stirrer at 3000 r/min to stir the material evenly, so that chain growth, gas generation and cross-linking reactions could be carried out almost simultaneously in a short time. Then, the mixture was quickly poured into the preheated mold, allowing free expansion and foaming for 1 min at room temperature. After the foam stopped growing, it was placed in an oven at 65 °C for 3 h to mature. Finally, the foam was placed at room temperature for 24 h until it was fully cured, and the properties of the polyurethane foaming materials were characterized. The ambient temperature was 10~20 °C, and the ambient humidity was 50~70%.
2.2.3. Observation of the Pore Morphology of Polyurethane Foam Materials
Scanning electron microscopy (SEM) 7610F and 7900 from Nippon Electronics Corporation were used. Before observation, the sample was placed on the sample table for gold-plating treatment. The gold-plated sample was placed under 10 kV acceleration voltage for observation, and the shooting was magnified 30 times.
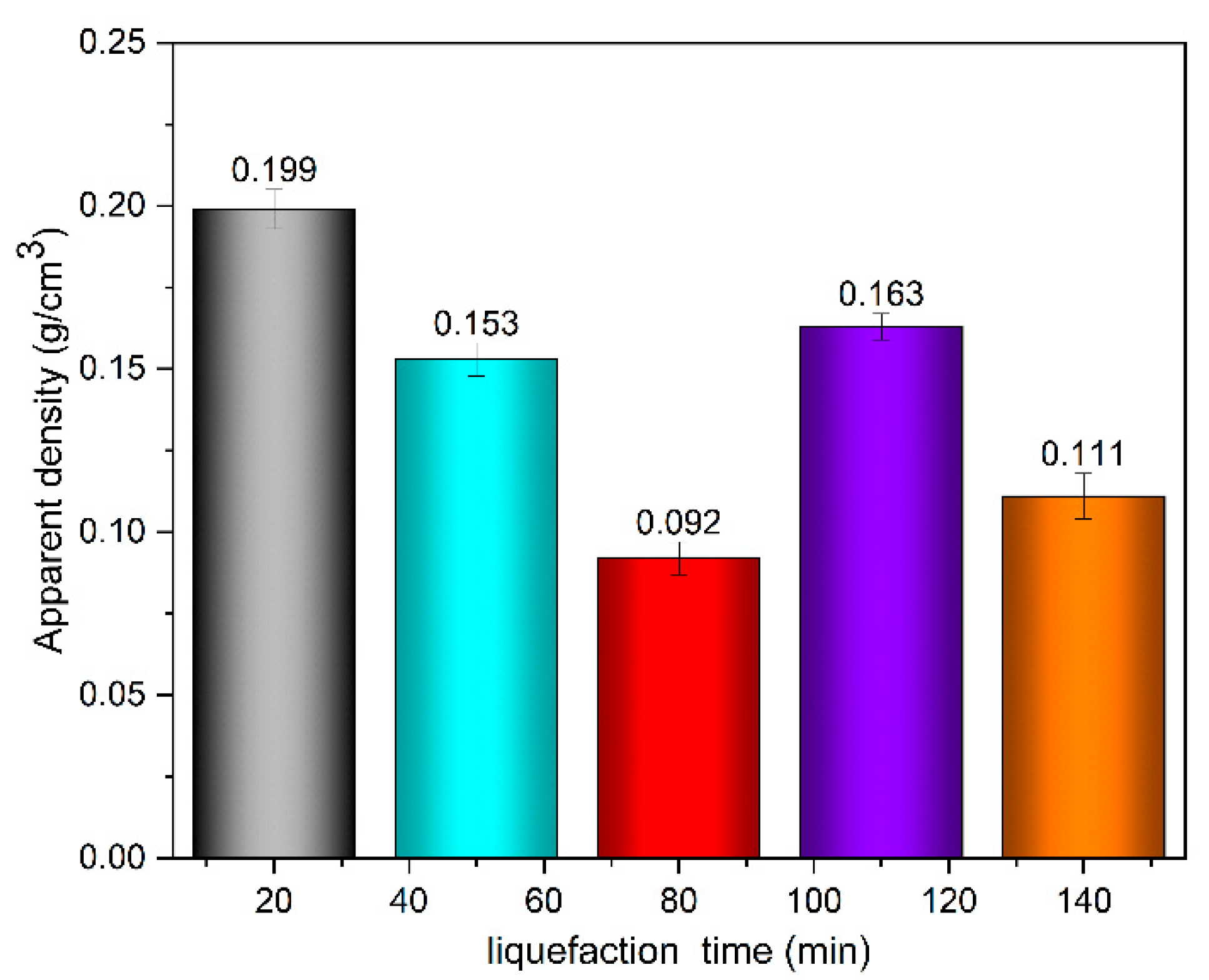
2.2.4. Determination of Density of Polyurethane Foaming Materials
Part of the properties of polyurethane foams can be described by the apparent density. The apparent density of the foam material was measured according to GB/T6343-2009 [
19]. The sample size was 30 mm × 30 mm × 30 mm, and the original bubble structure should not be destroyed when cutting. Five samples of each polyurethane foam material were made, and their mass was measured and recorded in g. The specific calculation method is as follows: take the average value of the five samples as the apparent density of polyurethane foam material, accurate to 0.1 kg /m
3.
2.2.5. Determination of Compression Properties of Polyurethane Foam Composites
A universal mechanical testing machine (Instron 3366, Instron, Boston, MA, USA) was used for measurement at room temperature according to ASTM D1621—16 standard [
20]. The sample size was 30 mm × 30 mm × 30 mm, and the compression speed was 5 mm/min. The compression strength and compression modulus of the material were read and recorded. Each group was repeated 5 times, and the average value was taken. Tests were conducted in the standard laboratory atmosphere of 23 °C ambient temperature and 50% relative humidity.
4. Conclusions
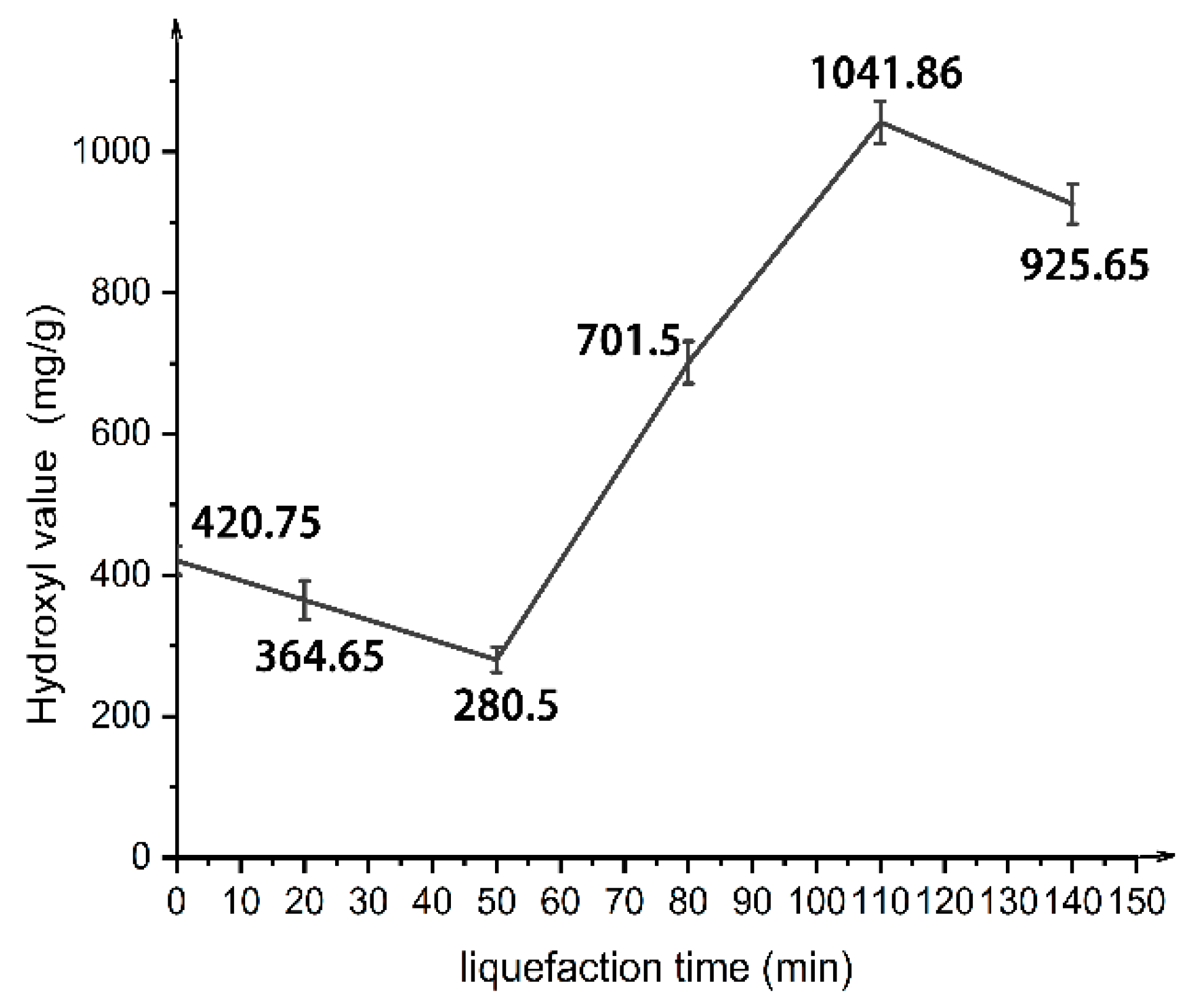
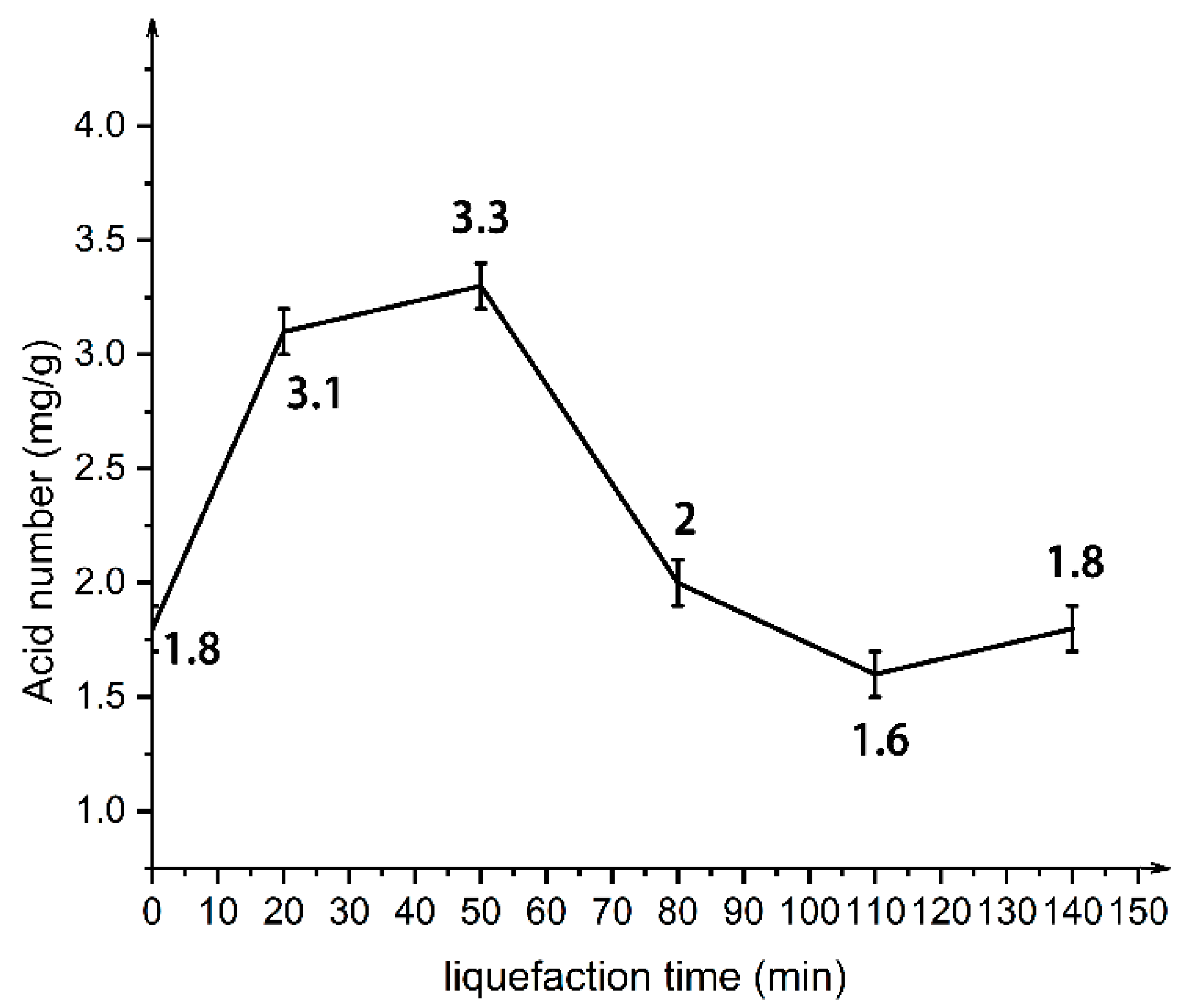
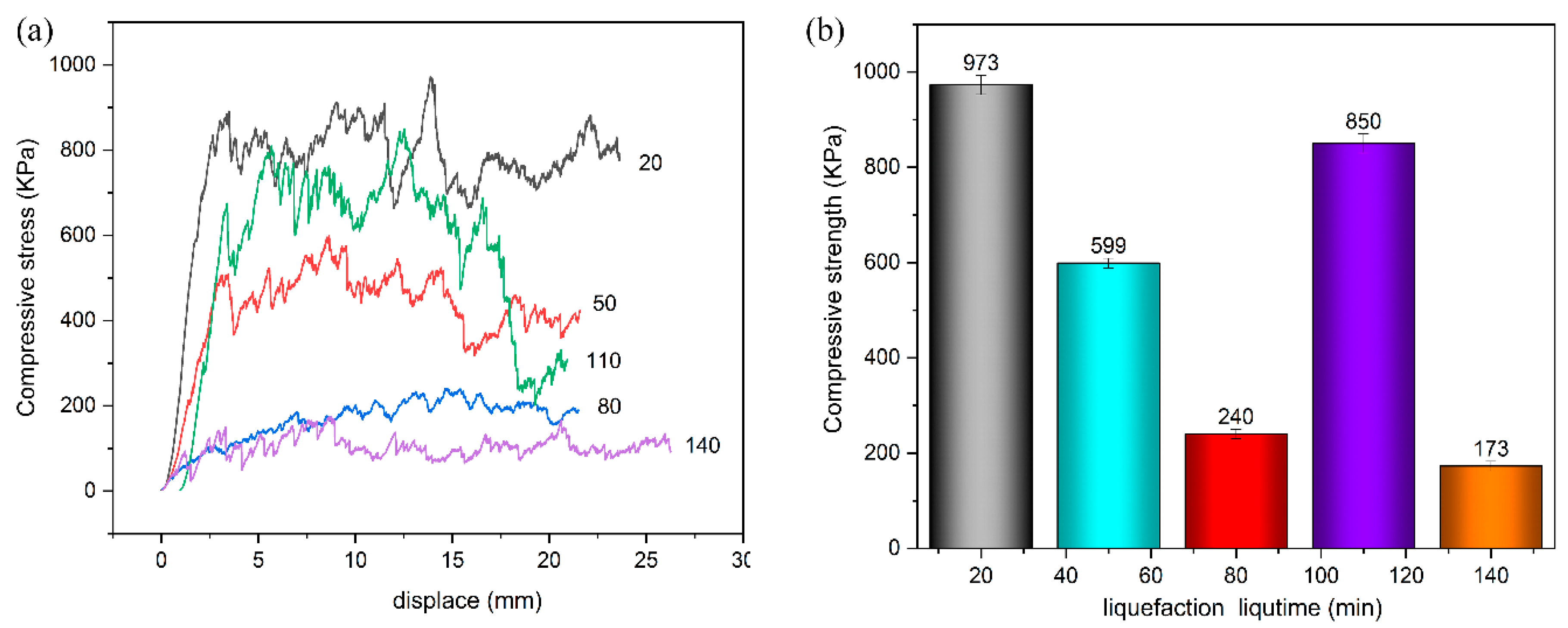
In this paper, by measuring the hydroxyl value of wood powder liquefaction products with different liquefaction time, infrared spectra, GPC spectra and 13C-NMR spectra, it was found that with the liquefaction time from 0 to 110 min, the hydroxyl value of wood powder liquefaction products first decreased and then increased. This is mainly because the polyol liquefaction of wood flour is a complex process, and different components of wood flour react differently at different stages. Moreover, it is also accompanied by the degradation reaction of biomass macromolecules, the solvation reaction between the liquefaction reagent polyhydroxy alcohol and the generated low molecular compound, as well as the condensation reaction between the generated low molecular compound (auto-condensation reaction) and the oxidation condensation reaction of the liquefaction agent. The hydroxyl value of the wood powder liquefaction product reached the maximum value of 1042 mg KOH/g when the liquefaction time was 110 min. Therefore, the optimal process time for the preparation of the wood powder liquefaction product, which could replace the traditional petroleum polyols, was 110 min. In addition, through the characterization of the morphology, apparent density and mechanical properties of polyurethane foam prepared by liquefaction products of wood powder with different liquefaction times, it was found that the polyurethane foam prepared by liquefaction products at 110 min had the smoothest surface, and the compression strength of 850 kPa was higher than that of 768 kPa of traditional polyurethane foam without wood powder; thus, the mechanical properties are good.
















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}