
3,4-Ethylenedioxythiophene (EDOT) End-Group Functionalized Poly-ε-caprolactone (PCL): Self-Assembly in Organic Solvents and Its Coincidentally Observed Peculiar Behavior in Thin Film and Protonated Media

 ,
, 
Abstract
: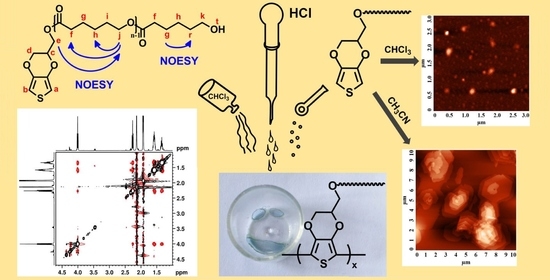
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Synthesis of EDOT-PCL Macromonomer
2.3. Measurements
3. Results and Discussion
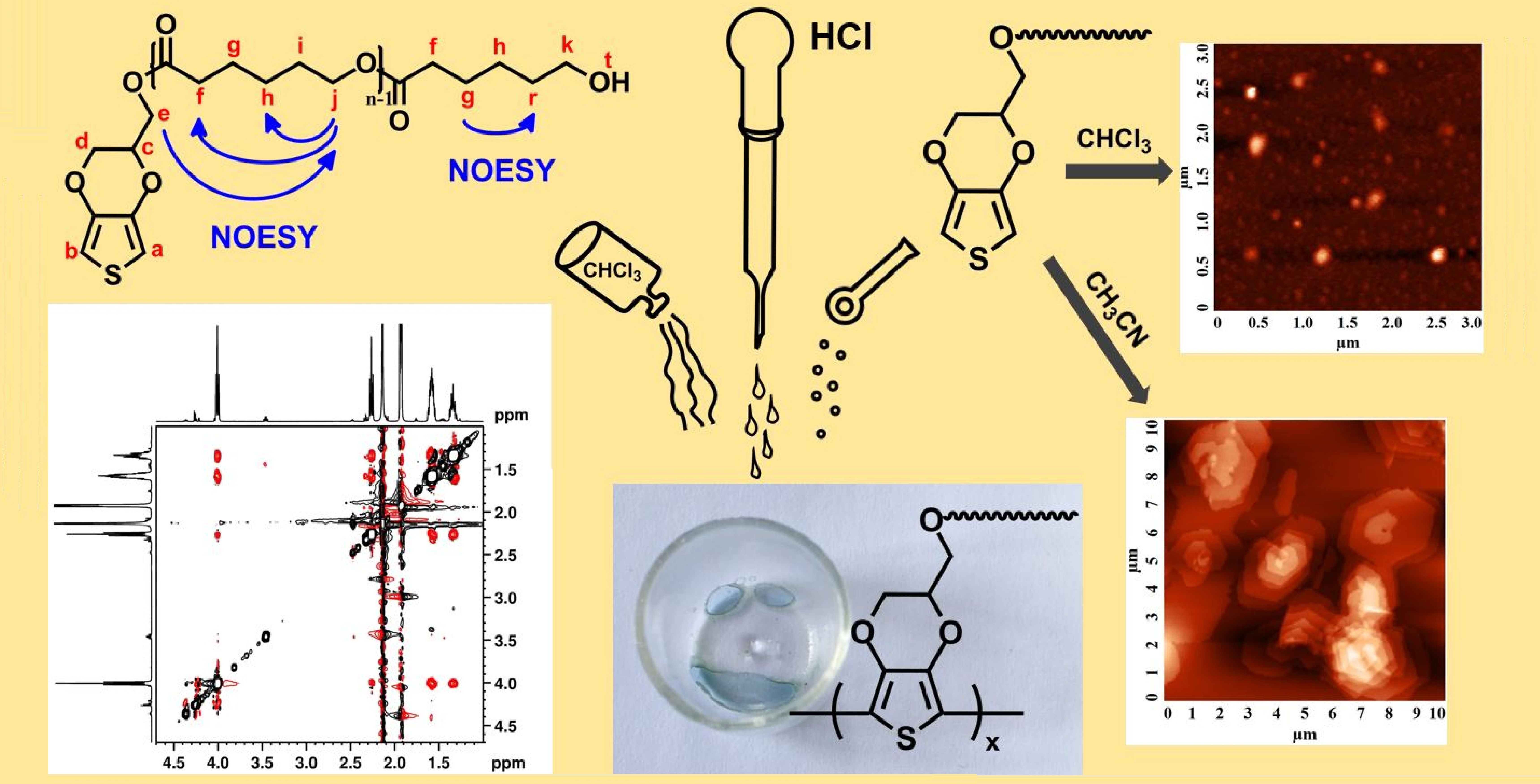
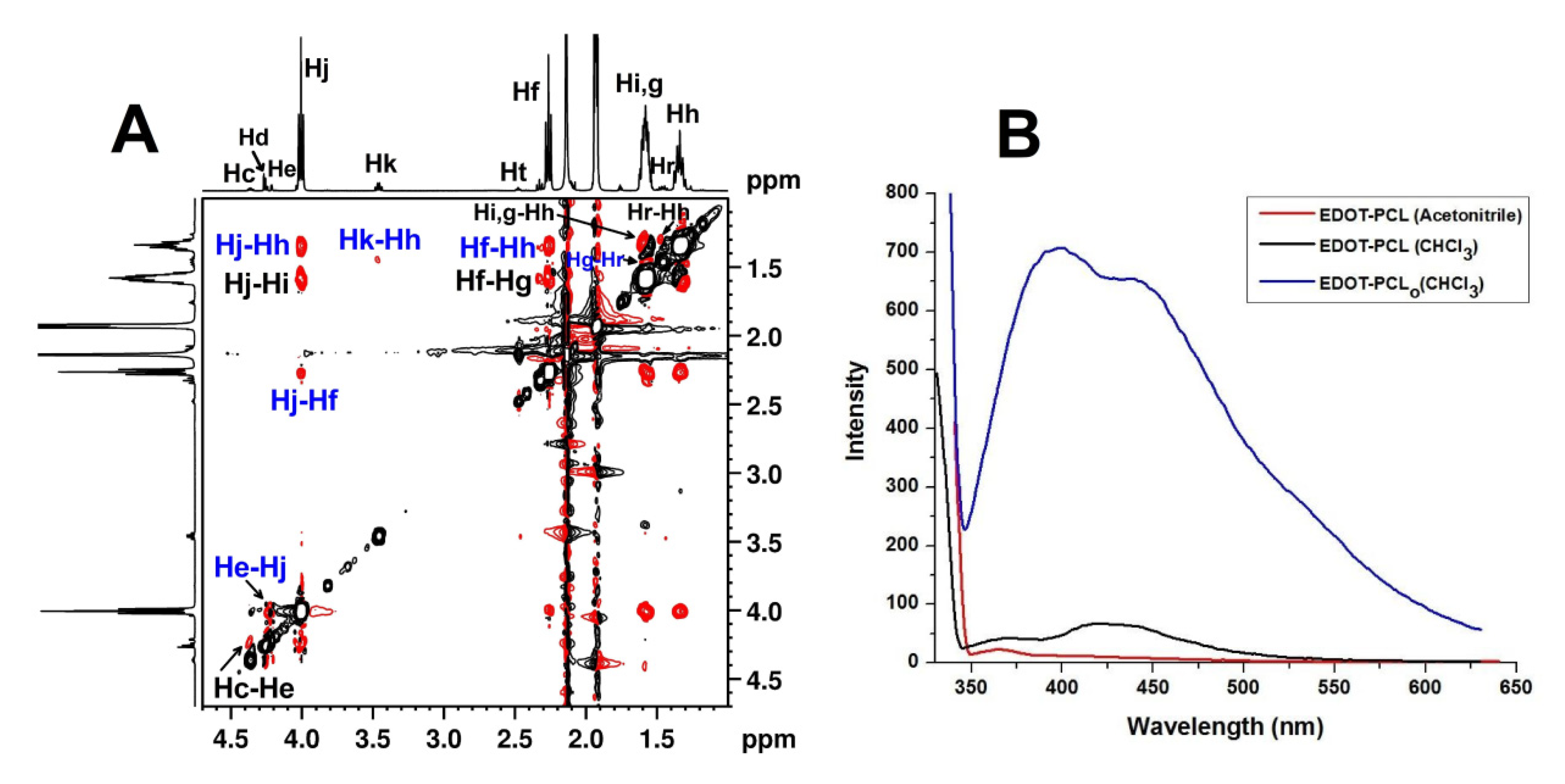
3.1. Behavior of EDOT-PCL in Solution
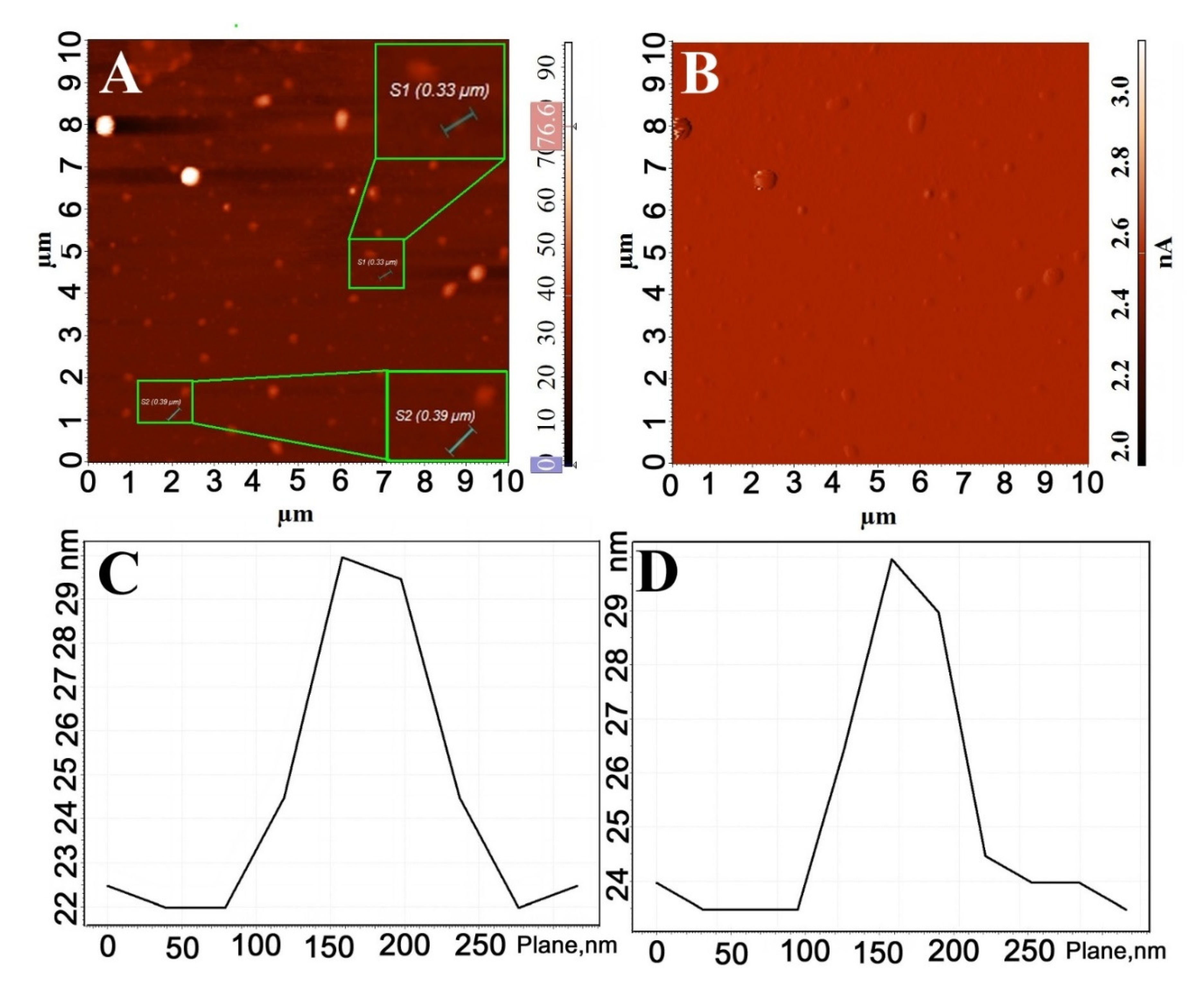
3.2. Surface Morphology Analysis of EDOT-PCL Thin Films
3.3. EDOT-PCL Behavior in the Presence of Acids
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Polymeropoulos, G.; Zapsas, G.; Ntetsikas, K.; Bilalis, P.; Gnanou, Y.; Hadjichristidis, N. 50th Anniversary Perspective: Polymers with Complex Architectures. Macromolecules 2017, 50, 1253–1290. [Google Scholar] [CrossRef]
- Lorenzo, A.T.; Muller, A.J.; Lin, M.-C.; Chen, H.-L.; Jeng, U.-S.; Priftis, D.; Pitsikalis, M.; Hadjichristidis, N. Influence of Macromolecular Architecture on the Crystallization of (PCL2)-b-(PS2) 4-Miktoarm Star Block Copolymers in Comparison to Linear PCL-b-PS Diblock Copolymer Analogues. Macromolecules 2009, 42, 8353–8364. [Google Scholar] [CrossRef]
- Hadjichristidis, N.; Pispas, S.; Pitsikalis, M. End-functionalized polymers with zwitterionic end-groups. Prog. Polym. Sci. 1999, 24, 875–915. [Google Scholar] [CrossRef]
- Lunn, D.J.; Discekici, E.H.; Read de Alaniz, J.; Gutekunst, W.R.; Hawker, C.J. Established and Emerging Strategies for Polymer Chain-End Modification. J. Polym. Sci. Part A Polym. Chem. 2017, 55, 2903–2914. [Google Scholar] [CrossRef] [Green Version]
- Liang, Y.; Deng, X.; Zhang, L.; Peng, X.; Gao, W.; Cao, J.; Gu, Z.; He, B. Terminal modification of polymeric micelles with π-conjugated moieties for efficient anticancer drug delivery. Biomaterials 2015, 71, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Rosberg, J.; Rottke, F.O.; Schulz, B.; Lendlein, A. Enzymatic Degradation of Oligo(ε-caprolactone)s End-Capped with Phenylboronic Acid Derivatives at the Air–Water Interface. Macromol. Rapid Commun. 2016, 37, 1966–1971. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Y.; Hadjichristidis, N. Tetraphenylethene-Functionalized Polyethylene-Based Polymers with Aggregation-Induced Emission. Macromolecules 2019, 52, 1955–1964. [Google Scholar] [CrossRef]
- Kim, J.; Jung, H.Y.; Park, M.J. End-Group Chemistry and Junction Chemistry in Polymer Science: Past, Present, and Future. Macromolecules 2020, 53, 746–763. [Google Scholar] [CrossRef] [Green Version]
- Cianga, I.; Yagci, Y. New polyphenylene-based macromolecular architectures by using well defined macromonomers synthesized via controlled polymerization methods. Prog. Polym. Sci. 2004, 29, 387–399. [Google Scholar] [CrossRef]
- Priya, M.H.; Pratt, L.R.; Paulaitis, M.E. Effect of PEG End-Group Hydrophobicity on Lysozyme Interactions in Solution Characterized by Light Scattering. Langmuir 2011, 27, 13713–13718. [Google Scholar] [CrossRef]
- Cheng, Y.; Hao, J.; Lee, L.A.; Biewer, M.C.; Wang, Q.; Stefan, M.C. Thermally Controlled Release of Anticancer Drug from Self-Assembled γ-Substituted Amphiphilic Poly(ε caprolactone) Micellar Nanoparticles. Biomacromolecules 2012, 13, 2163–2173. [Google Scholar] [CrossRef]
- Bendrea, A.-D.; Fabregat, G.; Cianga, L.; Estrany, F.; del Valle, L.J.; Cianga, I.; Aleman, C. Hybrid materials consisting of an all-conjugated polythiophene backbone and grafted hydrophilic poly(ethylene glycol) chains. Polym. Chem. 2013, 4, 2709–2723. [Google Scholar] [CrossRef]
- Dominguez-Alfaro, A.; Gabirondo, E.; Alegret, N.; De León-Almazán, C.M.; Hernandez, R.; Vallejo-Illarramendi, A.; Prato, M.; Mecerreyes, D. 3D Printable Conducting and Biocompatible PEDOT-graft-PLA Copolymers by Direct Ink Writing. Macromol. Rapid Commun. 2021, 2100100. [Google Scholar] [CrossRef]
- Cianga, I.; Mercore, V.M.; Grigoras, M.; Yagci, Y. Poly(thienyl-phenylene)s with Macromolecular Side Chains by Oxidative Polymerization of Well-Defined Macromonomers. J. Polym. Sci. Part A Polym. Chem. 2007, 45, 848–865. [Google Scholar] [CrossRef]
- Marina, S.; Mantione, D.; Manoj Kumar, K.; Kari, V.; Gutierrez, J.; Tercjak, A.; Sanchez-Sanchez, A.; Mecerreyes, D. New electroactive macromonomers and multi-responsive PEDOT graft copolymers. Polym. Chem. 2018, 9, 3780–3790. [Google Scholar] [CrossRef] [Green Version]
- Colak, D.G.; Cianga, I.; Muftoglu, A.E.; Yagci, Y. Synthesis and Characterization of Mid- and End-Chain Functional Telechelics by Controlled Polymerization Methods and Coupling Processes. J. Polym. Sci. Part A Polym. Chem. 2006, 44, 727–743. [Google Scholar] [CrossRef]
- Papila, O.; Toppare, L.; Yagci, Y.; Cianga, L. Conducting Copolymers of Thiophene- Functionalized Polystyrene. Int. J. Polym. Anal. Charact. 2004, 9, 13–28. [Google Scholar] [CrossRef]
- Scheinhardt, B.; Trzaskowski, J.; Baier, M.C.; Stempfle, B.; Oppermann, A.; Woll, D.; Mecking, S. Anisotropic Polyethylene Nanocrystals Labeled with a Single Fluorescent Dye Molecule: Toward Monitoring of Nanoparticle Orientation. Macromolecules 2013, 46, 7902–7910. [Google Scholar] [CrossRef]
- Demir, B.; Yilmaz, T.; Guler, E.; Pinar Gumus, Z.; Akbulut, H.; Aldemir, E.; Coskunol, H.; Colak, D.G.; Cianga, I.; Yamada, S.; et al. Polypeptide with electroactive endgroups as sensing platform for the abused drug ‘methamphetamine’ by bioelectrochemical method. Talanta 2016, 161, 789–796. [Google Scholar] [CrossRef]
- Li, C.Y. The rise of semicrystalline polymers and why are they still interesting. Polymer 2020, 211, 123150. [Google Scholar] [CrossRef]
- Sisson, A.L.; Ekinci, D.; Lendlein, A. The contemporary role of ε-caprolactone chemistry to create advanced polymer architectures. Polymer 2013, 54, 4333–4350. [Google Scholar] [CrossRef] [Green Version]
- Diaconescu, R.; Simionescu, B.C.; David, G. Control and prediction of degradation of biopolymer based hydrogels with poly( ε-caprolactone) subunits. Int. J. Biol. Macromol. 2014, 71, 147–154. [Google Scholar] [CrossRef]
- Simionescu, B.C.; Drobota, M.; Timpu, D.; Vasiliu, T.; Constantinescu, C.A.; Rebleanu, D.; Calin, M.; David, G. Biopolymers/poly(ε-caprolactone)/polyethylenimine functionalized nanohydroxyapatite hybrid cryogel: Synthesis, characterization and application in gene delivery. Mater. Sci. Eng. C 2017, 81, 167–176. [Google Scholar] [CrossRef] [PubMed]
- David, G.; Turin-Moleavin, I.; Ursu, L.-E.; Peptanariu, D.; Ailincai, D. Multilayer biopolymer/poly(ε-caprolactone)/polycation nanoparticles. Iran. Polym. J. 2018, 27, 517–526. [Google Scholar] [CrossRef]
- Ganda, S.; Dulle, M.; Drechsler, M.; Forster, B.; Forster, S.; Stenzel, M.H. Two-Dimensional Self-Assembled Structures of Highly Ordered Bioactive Crystalline-Based Block Copolymers. Macromolecules 2017, 50, 8544–8553. [Google Scholar] [CrossRef]
- Al-Sulami, A.; Ladelta, V.; Hadjichristidis, N. In-chain functionalized poly(ε-caprolactone): A valuable precursor towards the synthesis of 3-miktoarm star containing hyperbranched polyethylene. J. Polym. Sci. 2020, 58, 2764–2773. [Google Scholar] [CrossRef]
- Uyar, T.; Cianga, I.; Cianga, L.; Besenbacher, F.; Yagci, Y. Self-aligned and bundled electrospun fibers prepared from blends of polystyrene (PS) and poly(methyl methacrylate) (PMMA) with a hairy-rod polyphenylene copolymer. Mater. Lett. 2009, 63, 1638–1641. [Google Scholar] [CrossRef] [Green Version]
- Hardy, J.G.; Villancio-Wolter, M.K.; Sukhavasi, R.C.; Mouser, D.J.; Aguilar, D., Jr.; Geissler, S.A.; Kaplan, D.L.; Schmidt, C.E. Electrical Stimulation of Human Mesenchymal Stem Cells on Conductive Nanofibers Enhances their Differentiation toward Osteogenic Outcomes. Macromol. Rapid Commun. 2015, 36, 1884–1890. [Google Scholar] [CrossRef] [PubMed]
- Mei, S.; Staub, M.; Li, C.Y. Directed Nanoparticle Assembly through Polymer Crystallization. Chem. Eur. J. 2020, 26, 349–361. [Google Scholar] [CrossRef]
- Odrobinska, J.; Neugebauer, D. Micellar Carriers Based on Amphiphilic PEG/PCL Graft Copolymers for Delivery of Active Substances. Polymers 2020, 12, 2876. [Google Scholar] [CrossRef]
- Uyar, Z.; Durgun, M.; Sukru Yavuz, M.; Bahattin Abaci, M.; Arslan, U.; Degirmenci, M. Two-arm PCL and PLLA macrophotoinitiators with benzoin end-functional groups by combination of ROP and click chemistry and their use in the synthesis of A2B2 type miktoarm star copolymers. Polymer 2017, 123, 153–168. [Google Scholar] [CrossRef]
- Uyar, Z.; Cay, N.G.B.; Arslan, U.; Durgun, M.; Degirmenci, M. Synthesis and characterization of an A2B2-type miktoarm star copolymer based on poly(ε-caprolactone) and poly(cyclohexene oxide). Polym. Bull. 2019, 76, 553–573. [Google Scholar] [CrossRef]
- Degirmenci, M.; Izgin, O.; Yagci, Y. Synthesis and Characterization of Cyclohexene Oxide Functional Poly(ε-caprolactone) Macromonomers and Their Use in Photoinitiated Cationic Homo- and Copolymerization. J. Polym. Sci. Part A Polym. Chem. 2004, 42, 3365–3372. [Google Scholar] [CrossRef]
- Kang, E.-H.; Lee, I.-H.; Choi, T.-L. Brush Polymers Containing Semiconducting Polyene Backbones: Graft-Through Synthesis via Cyclopolymerization and Conformational Analysis on the Coil-to-Rod Transition. ACS Macro Lett. 2012, 1, 1098–1102. [Google Scholar] [CrossRef]
- Nikovia, C.; Sougioltzoupoulou, E.; Rigas, V.; Pitsikalis, M. Macromolecular Brushes Based on Poly(L-Lactide) and Poly(ε-Caprolactone) Single and Double Macromonomers via ROMP. Synthesis, Characterization and Thermal Properties. Polymers 2019, 11, 1606. [Google Scholar] [CrossRef] [Green Version]
- Hadjichristidis, N.; Pitsikalis, M.; Iatrou, H.; Pispas, S. The Strength of the Macromonomer Strategy for Complex Macromolecular Architecture: Molecular Characterization, Properties and Applications of Polymacromonomers. Macromol. Rapid Commun. 2003, 24, 979–1013. [Google Scholar] [CrossRef]
- Müllner, M.; Müller, A.H.E. Cylindrical polymer brushes—Anisotropic building blocks, unimolecular templates and particulate nanocarriers. Polymer 2016, 98, 389–401. [Google Scholar] [CrossRef]
- Xie, G.; Martinez, M.R.; Olszewski, M.; Sheiko, S.S.; Matyjaszewski, K. Molecular Bottlebrushes as Novel Materials. Biomacromolecules 2019, 20, 27–54. [Google Scholar] [CrossRef]
- Knaapila, M.; Stepanyan, R.; Lyons, B.P.; Torkkeli, M.; Monkman, A.P. Towards General Guidelines for Aligned, Nanoscale Assemblies of Hairy-Rod Polyfluorene. Adv. Funct. Mater. 2006, 16, 599–609. [Google Scholar] [CrossRef]
- Mecerreyes, D.; Pomposo, J.A.; Bengoetxea, M.; Grande, H. Novel Pyrrole End-Functional Macromonomers Prepared by Ring-Opening and Atom-Transfer Radical Polymerizations. Macromolecules 2000, 33, 5846–5849. [Google Scholar] [CrossRef]
- Molina, B.G.; Bendrea, A.D.; Cianga, L.; Armelin, E.; del Valle, L.J.; Cianga, I.; Alemán, C. The biocompatible polythiophene-g-polycaprolactone copolymer as an efficient dopamine sensor platform. Polym. Chem. 2017, 8, 6112–6122. [Google Scholar] [CrossRef] [Green Version]
- Molina, B.G.; Bendrea, A.-D.; Lanzalaco, S.; Franco, L.; Cianga, L.; del Valle, L.J.; Puiggali, J.; Turon, P.; Armelin, E.; Cianga, I.; et al. Smart design for a flexible, functionalized an electroresponsive hybrid platform based on poly(3,4-ethylenedioxythiophene) derivatives to improve cell viability. J. Mater. Chem. B 2020, 8, 8864–8877. [Google Scholar] [CrossRef]
- Molina, B.G.; Cianga, L.; Bendrea, A.-D.; Cianga, I.; Aleman, C.; Armelin, E. An amphiphilic, heterografted polythiophene copolymer containing biocompatible/ biodegradable side chains for use as an (electro) active surface in biomedical applications. Polym. Chem. 2019, 10, 5010–5022. [Google Scholar] [CrossRef]
- Chang, S.H.; Lee, H.J.; Park, S.; Kim, Y.; Jeong, B. Fast Degradable Polycaprolactone for Drug Delivery. Biomacromolecules 2018, 19, 2302–2307. [Google Scholar] [CrossRef] [PubMed]
- Tang, X.; Saveh-Shemshaki, N.; Kan, H.-M.; Khan, Y.; Laurencin, C.T. Biomimetic Electroconductive Nanofibrous Matrices for Skeletal Muscle Regenerative Engineering. Regen. Eng. Transl. Med. 2020, 6, 228–237. [Google Scholar] [CrossRef] [PubMed]
- Ritzau-Reid, K.I.; Spicer, C.D.; Gelmi, A.; Grigsby, C.L.; Ponder, J.F., Jr.; Bemmer, V.; Creamer, A.; Vilar, R.; Serio, A.; Stevens, M.M. An Electroactive Oligo-EDOT Platform for Neural Tissue Engineering. Adv. Funct. Mater. 2020, 30, 2003710. [Google Scholar] [CrossRef] [PubMed]
- Hollamby, M.J.; Karny, M.; Bomans, P.H.H.; Sommerdijk, N.A.J.M.; Saeki, A.; Seki, S.; Minamikawa, H.; Grillo, I.; Pauw, B.R.; Brown, P.; et al. Directed assembly of optoelectronically active alkyl–π-conjugated molecules by adding n-alkanes or π-conjugated species. Nat. Chem. 2014, 6, 690–696. [Google Scholar] [CrossRef]
- Zha, R.H.; de Waal, B.F.M.; Lutz, M.; Teunissen, A.J.P.; Meijer, E.W. End Groups of Functionalized Siloxane Oligomers Direct Block-Copolymeric or Liquid-Crystalline Self-Assembly Behavior. J. Am. Chem. Soc. 2016, 138, 5693–5698. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, X.; Xu, W.; Chang, X.; Zheng, Y.; Ni, L.; Shan, G.; Bao, Y.; Pan, P. Stepwise Crystallization and Induced Microphase Separation in Nucleobase Monofunctionalized Supramolecular Poly(ε-caprolactone). Macromolecules 2021, 54, 846–857. [Google Scholar] [CrossRef]
- Polarz, S.; Kunkel, M.; Donner, A.; Schlotter, M. Added-Value Surfactants. Chem. Eur. J. 2018, 24, 18842–18856. [Google Scholar] [CrossRef] [Green Version]
- Simionescu, C.I.; Grovu-Ivanoiu, M.; Cianga, I.; Grigoras, M.; Duca, A.; Cocarla, I. Electrochemical polymerization of some monomers with Schiff’s base structure. Angew. Makromol. Chem. 1996, 239, 1–12. [Google Scholar] [CrossRef]
- Pietrasik, J.; Sumerlin, B.S.; Lee, H.-I.; Gil, R.R.; Matyjaszewski, K. Structural mobility of molecular bottle-brushes investigated by NMR relaxation dynamics. Polymer 2007, 48, 496–501. [Google Scholar] [CrossRef]
- Cianga, L.; Sarac, A.; Ito, K.; Yagci, Y. Synthesis and Characterization of New Alternating, Amphiphilic, Comblike Copolymers of Poly(ethylene oxide) Macromonomer and N-Phenylmaleimide. J. Polym. Sci. Part A Polym. Chem. 2005, 43, 479–492. [Google Scholar] [CrossRef]
- Dissanayake, D.S.; Sheina, E.; Biewer, M.C.; McCullough, R.D.; Stefan, M.C. Determination of Absolute Molecular Weight of Regioregular Poly(3-Hexylthiophene) by 1H-NMR Analysis. J. Polym. Sci. Part A Polym. Chem. 2017, 55, 79–82. [Google Scholar] [CrossRef]
- Cianga, L.; Bendrea, A.-D.; Fifere, N.; Nita, L.E.; Doroftei, F.; Ag, D.; Seleci, M.; Timur, S.; Cianga, I. Fluorescent micellar nanoparticles by selfassembly of amphiphilic, nonionic and water selfdispersible polythiophenes with “hairy rod” architecture. RSC Adv. 2014, 4, 56385–56405. [Google Scholar] [CrossRef]
- Boccia, A.C.; Lukes, V.; Eckstein-Andicsova, A.; Kozma, E. Solvent- and concentration-induced self-assembly of an amphiphilic perylene dye. New J. Chem. 2020, 44, 892–899. [Google Scholar] [CrossRef]
- Abraham, R.J.; Mobli, M.; Smith, R.J. 1H chemical shifts in NMR: Part 19. Carbonyl anisotropies and steric effects in aromatic aldehydes and ketones. Magn. Reson. Chem. 2003, 41, 26–36. [Google Scholar] [CrossRef]
- Lomas, J.S. 1H NMR spectra of alcohols in hydrogen bonding solvents: DFT/GIAO calculations of chemical shifts. Magn. Reson. Chem. 2016, 54, 28–38. [Google Scholar] [CrossRef]
- Kontogianni, V.G.; Charisiadis, P.; Primikyri, A.; Pappas, C.G.; Exarchou, V.; Tzakos, A.G.; Gerothanassis, I.P. Hydrogen bonding probes of phenol—OH groups. Org. Biomol. Chem. 2013, 11, 1013–1025. [Google Scholar] [CrossRef]
- Zhou, Y.; Zheng, Y.-Z.; Sun, H.-Y.; Deng, G.; Yu, Z.-W. Hydrogen bonding interactions in ethanol and acetonitrile binary system: A near and mid-infrared spectroscopic study. J. Mol. Struct. 2014, 1069, 251–257. [Google Scholar] [CrossRef]
- Luo, C.J.; Stride, E.; Edirisinghe, M. Mapping the Influence of Solubility and Dielectric Constant on Electrospinning Polycaprolactone Solutions. Macromolecules 2012, 45, 4669–4680. [Google Scholar] [CrossRef]
- Boucher, D.S. Solubility parameters and solvent affinities for polycaprolactone: A comparison of methods. J. Appl. Polym. Sci. 2020, 137, 48908. [Google Scholar] [CrossRef]
- Liang, Y.; Sun, Y.; Fu, X.; Lin, Y.; Meng, Z.; Meng, Y.; Niu, J.; Lai, Y.; Sun, Y. The effect of π-Conjugation on the self-assembly of micelles and controlled cargo release. Artif. Cells Nanomed. Biotechnol. 2020, 48, 525–532. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mo, H.; Pochapsky, T.C. Intermolecular interactions characterized by nuclear Overhauser effects. Prog. Nucl. Magn. Reson. Spectrosc. 1997, 30, 1–38. [Google Scholar] [CrossRef]
- Voets, I.K.; De Keizer, A.; Cohen Stuart, M.A. Core and Corona Structure of Mixed Polymeric Micelles. Macromolecules 2006, 39, 5952–5955. [Google Scholar] [CrossRef]
- Landry, J.M.; Marangoni, D.G.; Lumsdern, M.D.; Berno, R. 1D and 2D NMR investigations of micelle-formation process in 8-phenyloctanoate micelles. Can. J. Chem. 2007, 85, 202–207. [Google Scholar] [CrossRef]
- Elgiddawy, N.; Ren, S.; Yassar, A.; Louis-Joseph, A.; Sauriat-Dorizon, H.; El Rouby, W.M.A.; El-Gendy, A.O.; Farghali, A.A.; Korri-Youssoufi, H. Dispersible Conjugated Polymer Nanoparticles as Biointerface Materials for Label-Free Bacteria Detection. ACS Appl. Mater. Interfaces 2020, 12, 39979–39990. [Google Scholar] [CrossRef]
- Hostnik, G.; Podlipnik, C.; Meriguet, G.; Cerar, J. Specificity of Counterion Binding to a Conjugated Polyelectrolyte: A Combined Molecular Dynamics and NOESY Investigation. Macromolecules 2020, 53, 1119–1128. [Google Scholar] [CrossRef] [Green Version]
- Hasan, N.; Fuchs, C.; Schwieger, C.; Busse, K.; Dolynchuk, O.; Kressler, J. Crystallization of poly(ε-caprolactone) at the air-water interface studied by IRRAS and GI-WAXS. Polymer 2020, 196, 122468. [Google Scholar] [CrossRef]
- Song, I.Y.; Kim, J.; Im, M.J.; Moon, B.J.; Park, T. Synthesis and Self-Assembly of Thiophene-Based All-Conjugated Amphiphilic Diblock Copolymers with a Narrow Molecular Weight Distribution. Macromolecules 2012, 45, 5058–5068. [Google Scholar] [CrossRef]
- Colak, D.G.; Cianga, I.; Cianga, L.; Yagci, Y. Synthesis and self-assembly of fluorenevinylene alternating copolymers in “Hairy-Rod” architecture: Side chain—mediated tuning of conformation, microstructure and photophysical properties. Des. Monomers Polym. 2016, 19, 508–534. [Google Scholar] [CrossRef] [Green Version]
- Bendrea, A.-D.; Cianga, L.; Hitruc, E.G.; Titorencu, I.; Cianga, I. Fluorescent Nanoparticles from “Hairy-Rods”, Water-Self Dispersible Amphiphilic Polythiophenes. Mater. Plast. 2013, 50, 71–78. [Google Scholar]
- Bendrea, A.-D.; Cianga, L.; Hitruc, E.G.; Cianga, I. Synthesis and Characterization of Fluorescent, Nonionic, Water Self-Dispersible Oligothiophenes. Int. J. Polym. Anal. Charact. 2013, 18, 189–198. [Google Scholar] [CrossRef]
- Casado, N.; Hernandez, G.; Veloso, A.; Devaraj, S.; Mecerreyes, D.; Armand, M. PEDOT Radical Polymer with Synergetic Redox and Electrical Properties. ACS Macro Lett. 2016, 5, 59–64. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, S.; Remita, H.; Ramos, L.; Dazzi, A.; Deniset-Besseau, A.; Beaunier, P.; Goubard, F.; Aubert, P.-H.; Brisset, F.; Remita, S. PEDOT nanostructures synthesized in hexagonal mesophases. New J. Chem. 2014, 38, 1106–1115. [Google Scholar] [CrossRef]
- Zambianchi, M.; Di Maria, F.; Cazzato, A.; Gigli, G.; Piacenza, M.; Della Sala, F.; Barbarella, G. Microwave-Assisted Synthesis of Thiophene Fluorophores, Labeling and Multilabeling of Monoclonal Antibodies, and Long Lasting Staining of Fixed Cells. J. Am. Chem. Soc. 2009, 131, 10892–10900. [Google Scholar] [CrossRef] [PubMed]
- Clark, A.P.-Z.; Shi, C.; Ng, B.C.; Wilking, J.N.; Ayzner, A.L.; Stieg, A.Z.; Schwartz, B.J.; Mason, T.G.; Rubin, Y.; Tolbert, S.H. Self-Assembling Semiconducting Polymers- Rods and Gels from Electronic Materials. ACS Nano 2013, 7, 962–977. [Google Scholar] [CrossRef]
- Poater, J.; Casanovas, J.; Sola, M.; Aleman, C. Examining the Planarity of Poly(3,4-ethylenedioxythiophene): Consideration of Self-Rigidification, Electronic, and Geometric Effects. J. Phys. Chem. A 2010, 114, 1023–1028. [Google Scholar] [CrossRef]
- Conboy, G.; Spencer, H.J.; Angioni, E.; Kanibolotsky, A.L.; Findlay, N.J.; Coles, S.J.; Wilson, C.; Pitak, M.B.; Risko, C.; Coropceanu, V.; et al. To bend or not to bend—are heteroatom interactions within conjugated molecules effective in dictating conformation and planarity? Mater. Horiz. 2016, 3, 333–339. [Google Scholar] [CrossRef] [Green Version]
- Bendrea, A.-D.; Cianga, L.; Cianga, I. Poly(ethylene glycol)- functionalized Water Self-Dispersible α-Terthiophenes. Rev. Roum. Chim. 2013, 58, 153–160. [Google Scholar]
- Rayeroux, D.; Travelet, C.; Lapinte, V.; Borsali, R.; Robin, J.-J.; Bouilhac, C. Tunable amphiphilic graft copolymers bearing fatty chains and polyoxazoline: Synthesis and self-assembly behavior in solution. Polym. Chem. 2017, 8, 4246–4263. [Google Scholar] [CrossRef]
- Xiang, L.; Ryu, W.; Kim, H.; Ree, M. Precise Synthesis, Properties, and Structures of Cyclic Poly(ε-caprolactone)s. Polymers 2018, 10, 577. [Google Scholar] [CrossRef] [Green Version]
- Rodrigues-Ropero, F.; Casanovas, J.; Aleman, C. Ab Initio Calculations on π -Stacked Thiophene Dimer, Trimer, and Tetramer: Structure, Interaction Energy, Cooperative Effects, and Intermolecular Electronic Parameters. J. Comput. Chem. 2008, 29, 69–78. [Google Scholar] [CrossRef]
- Guo, C.; Huo, G. Poor solvent as a nucleating agent to induce poly(ε-caprolactone) ultrathin film crystallization on poly(vinylpyrrolidone) substrate. Colloid Polym. Sci. 2016, 294, 767–776. [Google Scholar] [CrossRef]
- Zhao, J.; Zhang, J.; Duan, X.; Peng, Z.; Wang, S. Formation mechanism, chain folding, and growth behavior of the intriguing fiber-like crystal of poly (ethylene oxide-b-ε-caprolactone) block copolymer in ultra thin films. Polymer 2011, 52, 2085–2093. [Google Scholar] [CrossRef]
- Huang, Y.; Liu, X.-B.; Zhang, H.-L.; Zhu, D.-S.; Sun, Y.-J.; Yan, S.-K.; Wang, J.; Chen, X.-F.; Wan, X.-H.; Chen, E.-Q.; et al. AFM study of crystallization and melting of a poly(ethylene oxide) diblock copolymer containing a tablet-like block of poly{2,5-bis[(4-methoxyphenyl)oxycarbonyl]styrene} in ultra thin films. Polymer 2006, 47, 1217–1225. [Google Scholar] [CrossRef]
- Wang, J.; Li, L.; Yang, W.; Yan, Z.; Zhou, Y.; Wang, B.; Zhang, B.; Bu, W. Sub-10 nm Scale Lamellar Structures with a High Degree of Long-Range Order Fabricated by Orthogonal Self-Assembly of Crown Ether/Secondary Dialkylammonium Recognition and Metal···Metal/ π−π Interactions. ACS Macro Lett. 2019, 8, 1012–1016. [Google Scholar] [CrossRef]
- Levent Demirel, A.; Yurteri, S.; Cianga, I.; Yagci, Y. Layered Morphology of Poly(phenylene)s in Thin Films Induced by Substitution of Well-Defined Poly(ε-caprolactone) Side Chains. Macromolecules 2005, 38, 6402–6410. [Google Scholar] [CrossRef]
- He, W.-N.; Xu, J.-T. Crystallization assisted self-assembly of semicrystalline block copolymers. Prog. Polym. Sci. 2012, 37, 1350–1400. [Google Scholar] [CrossRef]
- Crassous, J.J.; Schurtenberger, P.; Ballauff, M.; Mihut, A.M. Design of block copolymer micelles via crystallization. Polymer 2015, 62, A1–A13. [Google Scholar] [CrossRef]
- Van Horn, R.M.; Steffen, M.R.; O’Connor, D. Recent progress in block copolymer crystallization. Polym. Crystall. 2018, 1, e10039. [Google Scholar] [CrossRef]
- Michell, R.M.; Müller, A.J. Confined crystallization of polymeric materials. Prog. Polym. Sci. 2016, 54–55, 183–213. [Google Scholar] [CrossRef]
- Liu, Y.-X.; Chen, E.-Q. Polymer crystallization of ultrathin films on solid substrates. Coord. Chem. Rev. 2010, 254, 1011–1037. [Google Scholar] [CrossRef]
- Ganda, S.; Stenzel, M.H. Concepts, fabrication methods and applications of living crystallization-driven self-assembly of block copolymers. Prog. Polym. Sci. 2020, 101, 101195. [Google Scholar] [CrossRef]
- Cha, Y.; Jarrett-Wilkins, C.; Rahman, M.A.; Zhu, T.; Sha, Y.; Manners, I.; Tang, C. Crystallization-Driven Self-Assembly of Metallo-Polyelectrolyte Block Copolymers with a Polycaprolactone Core-Forming Segment. ACS Macro Lett. 2019, 8, 835–840. [Google Scholar] [CrossRef]
- Rejek, T.; Schweizer, P.; Joch, D.; Portilla, L.; Spiecker, E.; Halik, M. Buried Microphase Separation by Dynamic Interplay of Crystallization and Microphase Separation in Semicrystalline PEO Rich PS-b-PEO Block Copolymer Thin Films. Macromolecules 2020, 53, 5604–5613. [Google Scholar] [CrossRef]
- Wei, Y.; Pan, C.; Li, B.; Han, Y. Self-assembly morphology effects on the crystallization of semicrystalline block copolymer thin film. J. Chem. Phys. 2007, 126, 104902. [Google Scholar] [CrossRef]
- Di Maria, F.; Fabiano, E.; Gentili, D.; Biasiucci, M.; Salzillo, T.; Bergamini, G.; Gazzano, M.; Zanelli, A.; Brillante, A.; Cavallini, M.; et al. Polymorphism in Crystalline Microfibers of Achiral Octithiophene: The Effect on Charge Transport, Supramolecular Chirality and Optical Properties. Adv. Funct. Mater. 2014, 24, 4943–4951. [Google Scholar] [CrossRef]
- Vleugels, M.E.J.; de Zwart, M.E.; Magana, J.R.; Lamers, B.A.G.; Voets, I.K.; Meijer, E.W.; Petkau-Milroy, K.; Palmans, A.R.A. Effects of crystallinity and dispersity on the self-assembly behavior of block co-oligomers in water. Polym. Chem. 2020, 11, 7170–7177. [Google Scholar] [CrossRef]
- Doncom, K.E.B.; Blackman, L.D.; Wright, D.B.; Gibson, M.I.; O’Reilly, R.K. Dispersity effects in polymer self-assemblies: A matter of hierarchical control. Chem. Soc. Rev. 2017, 46, 4119–4134. [Google Scholar] [CrossRef] [Green Version]
- Wang, G.; Jiang, C.; Gao, C.; Wang, X.; Chen, G. Dispersion of Electrically Conductive Polymer PEDOT in Organic Solvents. Acta Polym. Sin. 2014, 11, 1532–1538. [Google Scholar] [CrossRef]
- Kawazu, K.; Nakagawa, S.; Ishizone, T.; Nojima, S. Effects of Bulky End-Groups on the Crystallization Kinetics of Poly(ε-caprolactone) Homopolymers Confined in a Cylindrical Nanodomain. Macromolecules 2017, 50, 7202–7210. [Google Scholar] [CrossRef]
- Zhang, L.; Wang, T.; Shen, Z.; Liu, M. Chiral Nanoarchitectonics: Towards the Design, Self-Assembly, and Function of Nanoscale Chiral Twists and Helices. Adv. Mater. 2016, 28, 1044–1059. [Google Scholar] [CrossRef]
- Song, B.; Liu, B.; Jin, Y.; He, X.; Tang, D.; Wu, G.; Yin, S. Controlled self-assembly of helical nano-ribbons formed by achiral amphiphiles. Nanoscale 2015, 7, 930–935. [Google Scholar] [CrossRef]
- Lee, E.; Hammer, B.; Kim, J.-K.; Page, Z.; Emrick, T.; Hayward, R.C. Hierarchical Helical Assembly of Conjugated Poly(3-hexylthiophene)-block-poly(3-triethylene glycol thiophene) Diblock Copolymers. J. Am. Chem. Soc. 2011, 133, 10390–10393. [Google Scholar] [CrossRef] [PubMed]
- Cui, Y.; Tao, D.; Huang, X.; Lu, G.; Feng, C. Self-Assembled Helical and Twisted Nanostructures of a Preferred Handedness from Achiral π-Conjugated Oligo(p-phenylenevinylene) Derivatives. Langmuir 2019, 35, 3134–3142. [Google Scholar] [CrossRef] [PubMed]
- Zhong, S.; Cui, H.; Chen, Z.; Wooley, K.L.; Pochan, D.J. Helix self-assembly through the coiling of cylindrical micelles. Soft Matter 2008, 4, 90–93. [Google Scholar] [CrossRef]
- Sanandaji, N.; Ovaskainen, L.; Gunnewiek, M.K.; Vancso, G.J.; Hedenqvist, M.S.; Yu, S.; Eriksson, L.; Roth, S.V.; Gedde, U.W. Unusual crystals of poly(ε-caprolactone) by unusual crystallisation: The effects of rapid cooling and fast solvent loss on the morphology, crystal structure and melting. Polymer 2013, 54, 1497–1503. [Google Scholar] [CrossRef]
- Iwata, T.; Furuhashi, Y.; Su, F.; Doi, Y. Single crystal morphologies of biodegradable aliphatic polyesters. Riken Rev. 2001, 42, 15–18. [Google Scholar]
- Iwata, T.; Doi, Y. Morphology and enzymatic degradation of poly(ε-caprolactone) single crystals: Does a polymer single crystal consist of micro-crystals? Polym. Int. 2002, 51, 852–858. [Google Scholar] [CrossRef]
- Zhang, H.; Liu, M.; Zhou, T.; Dong, B.; Li, C.Y. Stepwise assembly of a cross-linked free-standing nanoparticle sheet with controllable shape. Nanoscale 2015, 7, 11033–11039. [Google Scholar] [CrossRef] [PubMed]
- Bittinger, H.; Marchessault, R.H.; Niegisch, W.D. Crystal Structure of Poly-ε-caprolactone. Acta Cryst. 1970, B26, 1923–1927. [Google Scholar] [CrossRef]
- Su, M.; Huang, H.; Ma, X.; Wang, Q.; Su, Z. Poly(2-vinylpyridine)-block-Poly(ε-caprolactone) Single Crystals in Micellar Solution. Macromol. Rapid Commun. 2013, 34, 1067–1071. [Google Scholar] [CrossRef] [PubMed]
- Mareau, V.H.; Prud’Homme, R.E. In-Situ Hot Stage Atomic Force Microscopy Study of Poly(ε-caprolactone) Crystal Growth in Ultrathin Films. Macromolecules 2005, 38, 398–408. [Google Scholar] [CrossRef]
- Beekmans, L.G.M.; Vancso, G.J. Real-time crystallization study of poly(e-caprolactone) by hot-stage atomic force microscopy. Polymer 2000, 41, 8975–8981. [Google Scholar] [CrossRef]
- Qi, H.; Wang, W.; Li, C.Y. Janus Polymer Single Crystal Nanosheet via Evaporative Crystallization. ACS Macro Lett. 2014, 3, 675–678. [Google Scholar] [CrossRef]
- Nunez, E.; Gedde, U.W. Single crystal morphology of star-branched polyesters with crystallisable poly(3-caprolactone) arms. Polymer 2005, 46, 5992–6000. [Google Scholar] [CrossRef]
- Lotz, B.; Wittmann, J.C. Structure of Polymer Single Crystals in Materials Science and Technology, Structure and Properties of Polymers; Cahn, R.W., Haasen, P., Kramer, E.J., Eds.; VCH: Weinheim, Germany, 1993; Volume 12, pp. 79–151, Chapter 3. [Google Scholar]
- Diaz, A.; Bacaicoa, A.; Casas, M.T.; Franco, L.; Serra, A.; Puiggali, J. Study on the crystallization of multiarm stars with a poly (ethyleneimine) core and poly(ε-caprolactone) arms of different length. Thermochim. Acta 2015, 607, 39–52. [Google Scholar] [CrossRef]
- Agbolaghi, S.; Abbaspoor, S.; Abbasi, F. A comprehensive review on polymer single crystals—From fundamental concepts to applications. Prog. Polym. Sci. 2018, 81, 22–79. [Google Scholar] [CrossRef]
- Nunez, E.; Vancso, G.J.; Gedde, U.W. Morphology, Crystallization, and Melting of Single Crystals and Thin Films of Star-branched Polyesters with Poly(ε-caprolactone) Arms as Revealed by Atomic Force Microscopy. J. Macromol. Sci. Part B Physics 2008, 47, 589–607. [Google Scholar] [CrossRef]
- Armelin, E.; Almontassir, A.; Franco, L.; Puiggali, J. Crystalline Structure of Poly(decamethylene sebacate). Repercussions on Lamellar Folding Surfaces. Macromolecules 2002, 35, 3630–3635. [Google Scholar] [CrossRef]
- Sanandaji, N.; Neway, B.; Hedenqvist, M.S.; Li, Y.; Hawker, C.J.; Takizawa, K.; Gedde, U. Crystallization and Melting Behavior of Monodisperse Oligomers of ε-Caprolactone. J. Macromol. Sci. Part B Physics 2012, 51, 2075–2092. [Google Scholar] [CrossRef]
- Vasiliev, C.; Gunter, R.; Pispas, P.; Hadjichristidis, N. Crystallization pf block copolymers in restricted cylindrical geometries. Polymer 2006, 47, 330–340. [Google Scholar] [CrossRef]
- Huang, Y.; Mai, Y.; Yang, X.; Beser, U.; Liu, J.; Zhang, F.; Yan, D.; Mullen, K.; Feng, X. Temperature-Dependent Multidimensional Self-Assembly of Polyphenylene-Based “Rod−Coil” Graft Polymers. J. Am. Chem. Soc. 2015, 137, 11602–11605. [Google Scholar] [CrossRef] [PubMed]
- Simpson, J.H. Organic Structure Determination Using 2-D NMR Spectrocopy- A Problem-Based Approach, 2nd ed.; Ch. 2—“Instrumental Considerations”; Elsevier: Amsterdam, The Netherlands, 2012; pp. 21–57. [Google Scholar]
- Elschner, A.; Kirchmeyer, S.; Lovenich, W.; Merker, U.; Reuter, K. PEDOT—Principle and Applications of an Intrinsically Conductive Polymer; ch. 5—“The Synthesis of EDOT Monomer and Its Physical and Chemical Properties”; CRC Press: Boca Raton, FL, USA, 2011; pp. 47–66. [Google Scholar]
- Zhang, S.; Zhang, W.; Zhang, G.; Bai, Y.; Chen, S.; Xu, J.; Yu, Z.; Sun, K. p-Toluenesulfonic acid catalytic polymerization of EDOT without oxidants. Mater. Lett. 2018, 222, 105–108. [Google Scholar] [CrossRef]
- Tomsik, E.; Ivanko, I.; Svoboda, J.; Sedenkova, I.; Zhigunov, A.; Hromadkova, J.; Panek, J.; Lukesova, M.; Velychkivska, N.; Janisova, L. Method of Preparation of Soluble PEDOT: Self-Polymerization of EDOT without Oxidant at Room Temperature. Macromol. Chem. Phys. 2020, 221, 2000219. [Google Scholar] [CrossRef]
- Elschner, A.; Kirchmeyer, S.; Lovenich, W.; Merker, U.; Reuter, K. PEDOT—Principle and Applications of an Intrinsically Conductive Polymer; ch. 6—“From EDOT to PEDOT: Oxidative Polymerization and Other Routes”; CRC Press: Boca Raton, FL, USA, 2011; pp. 67–82. [Google Scholar]
- Cho, W.; Wu, J.; Shim, B.S.; Kuan, W.-F.; Mastroianni, S.E.; Young, W.-S.; Kuo, C.-C.; Epps, T.H., III; Martin, D.C. Synthesis and characterization of bicontinuous cubic poly(3,4-ethylene dioxythiophene) gyroid (PEDOT GYR) gels. Phys. Chem. Chem. Phys. 2015, 17, 5115–5123. [Google Scholar] [CrossRef] [Green Version]
- Qin, L.; Ding, Z.; Hanif, M.; Jiang, J.; Liu, L.; Mo, Y.; Xie, Z.; Ma, Y. Poly(3,4-dioxythiophene) soft nano-network with a compatible ion transporting channel for improved electrochromic performance. Polym. Chem. 2016, 7, 6954–6963. [Google Scholar] [CrossRef]
- Zhao, Q.; Jamal, R.; Zhang, L.; Wang, M.; Abdiryim, T. The structure and properties of PEDOT synthesized by template-free solution method. Nanoscale Res. Lett. 2014, 9, 557. [Google Scholar] [CrossRef] [Green Version]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bendrea, A.-D.; Cianga, L.; Ailiesei, G.-L.; Ursu, E.-L.; Göen Colak, D.; Cianga, I. 3,4-Ethylenedioxythiophene (EDOT) End-Group Functionalized Poly-ε-caprolactone (PCL): Self-Assembly in Organic Solvents and Its Coincidentally Observed Peculiar Behavior in Thin Film and Protonated Media. Polymers 2021, 13, 2720. https://doi.org/10.3390/polym13162720
Bendrea A-D, Cianga L, Ailiesei G-L, Ursu E-L, Göen Colak D, Cianga I. 3,4-Ethylenedioxythiophene (EDOT) End-Group Functionalized Poly-ε-caprolactone (PCL): Self-Assembly in Organic Solvents and Its Coincidentally Observed Peculiar Behavior in Thin Film and Protonated Media. Polymers. 2021; 13(16):2720. https://doi.org/10.3390/polym13162720
Chicago/Turabian StyleBendrea, Anca-Dana, Luminita Cianga, Gabriela-Liliana Ailiesei, Elena-Laura Ursu, Demet Göen Colak, and Ioan Cianga. 2021. "3,4-Ethylenedioxythiophene (EDOT) End-Group Functionalized Poly-ε-caprolactone (PCL): Self-Assembly in Organic Solvents and Its Coincidentally Observed Peculiar Behavior in Thin Film and Protonated Media" Polymers 13, no. 16: 2720. https://doi.org/10.3390/polym13162720