Effect of Stereolithography 3D Printing on the Properties of PEGDMA Hydrogels



Abstract
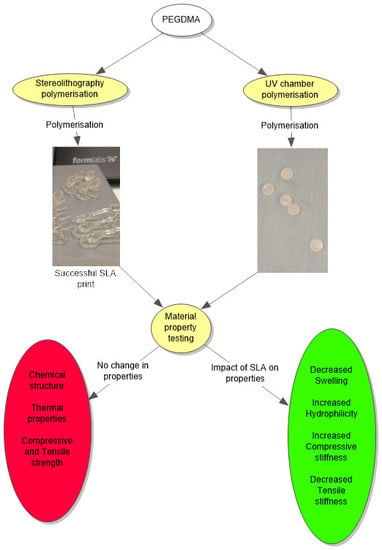
:
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Hydrogel Fabrication
2.3. Material Property Characterisation
2.3.1. Swelling Studies
2.3.2. Chemical Analysis
2.3.3. Dynamic Mechanical Analysis
2.3.4. Compression Testing
2.3.5. Wettability Measurement
2.3.6. Thermal Properties
2.3.7. Statistical Analysis
3. Results
3.1. Swelling Characteristics
3.2. Chemical Analysis
3.3. Dynamic Mechanical Analysis
3.4. Compression Testing
3.5. Wettability Measurements
3.6. Thermal Properties
4. Discussion
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Stoddard, S.D.; Alamos, D.; Harper, T.; Village, I.; Commission, A.E. Process of Photopolymerisation. U.S. Patent 2,367,661, 23 January 1945. [Google Scholar]
- Stoddard, S.D.; Alamos, D.; Harper, T.; Village, I.; Commission, A.E. Solvent Development of Photopolymerised Layers. U.S. Patent 3,475,171, 28 October 1969. [Google Scholar]
- Stoddard, S.D.; Alamos, D.; Harper, T.; Village, I.; Commission, A.E. Treatment of Diazo-Sensitised Lithographic Plates. U.S. Patent 3,019,105, 30 January 1962. [Google Scholar]
- Martin, E.L. Photopolymerisable Compositions and Elements and Process of Making Reliefs Therefrom. U.S. Patent 2,902,365, 1 September 1959. [Google Scholar]
- O’Brien, A.K.; Bowman, C.N. Impact of oxygen on photopolymerization kinetics and polyme structure. Macromolecules 2006, 39, 2501–2506. [Google Scholar] [CrossRef]
- Bowman, C.N.; Kloxin, C.J. Toward an Enhanced Understanding and Implementation of Photopolymerization Reactions. Am. Inst. Chem. Eng. 2008, 54, 2775–2795. [Google Scholar] [CrossRef]
- Billiet, T.; Vandenhaute, M.; Schelfhout, J.; Van Vlierberghe, S.; Dubruel, P. A review of trends and limitations in hydrogel-rapid prototyping for tissue engineering. Biomaterials 2012, 33, 6020–6041. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Goyanes, A.; Gaisford, S.; Basit, A.W. Stereolithographic (SLA) 3D printing of oral modified-release dosage forms. Int. J. Pharm. 2016, 503, 207–212. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.; Yeatts, A.; Dean, D.; Fisher, J.P. Stereolithographic bone scaffold design parameters: Osteogenic differentiation and signal expression. Tissue Eng. Part B Rev. 2010, 16, 523–539. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- PICO2 HD-Products-Asiga. Available online: https://www.asiga.com/products/printers/pico2_series/pico2hd/ (accessed on 5 August 2018).
- Annabi, N.; Nichol, J.W.; Zhong, X.; Ji, C.; Koshy, S.T.; Khademhosseini, A.; Dehghani, F. Controlling the Porosity and Microarchitecture of Hydrogels for Tissue Engineering. Tissue Eng. Part B Rev. 2010, 16, 371–383. [Google Scholar] [CrossRef]
- Peppas, N.A.; Hilt, J.Z.; Khademhosseini, A.; Langer, R. Hydrogels in biology and medicine: From molecular principles to bionanotechnology. Adv. Mater. 2006, 18, 1345–1360. [Google Scholar] [CrossRef]
- Fuenmayor, E.; Forde, M.; Healy, A.V.; Devine, D.M.; Lyons, J.G.; McConville, C.; Major, I. Comparison of fused-filament fabrication to direct compression and injection molding in the manufacture of oral tablets. Int. J. Pharm. 2019, 558, 328–340. [Google Scholar] [CrossRef] [Green Version]
- Major, I.; Fuenmayor, E.; McConville, C. The Production of Solid Dosage Forms from Non-Degradable Polymers. Curr. Pharm. Des. 2016, 22, 2738–2760. [Google Scholar] [CrossRef]
- Arcaute, K.; Mann, B.K.; Wicker, R.B. Stereolithography of three-dimensional bioactive poly(ethylene glycol) constructs with encapsulated cells. Ann. Biomed. Eng. 2006, 34, 1429–1441. [Google Scholar] [CrossRef]
- Mendes-Felipe, C.; Patrocinio, D.; Laza, J.M.; Ruiz-Rubio, L.; Vilas-Vilela, J.L. Evaluation of postcuring process on the thermal and mechanical properties of the Clear02TM resin used in stereolithography. Polym. Test. 2018, 72, 115–121. [Google Scholar] [CrossRef]
- Hutmacher, D.; Woodfield, T.; Dalton, P.; Lewis, J. Scaffold design and fabrication. Tissue Eng. 2008, 403–454. [Google Scholar]
- O’Brien, F.J. Biomaterials & scaffolds for tissue engineering. Mater. Today 2011, 14, 88–95. [Google Scholar]
- Kwan, M.K.; Wall, E.J.; Massie, J.; Garfin, S.R. Strain, stress and stretch of peripheral nerve. Rabbit experiments in vitro and in vivo. Acta Orthop. Scand. 1992, 63, 267–272. [Google Scholar] [CrossRef] [PubMed]
- Kerin, A.J.; Wisnom, M.R.; Adams, M.A. The compressive strength of articular cartilage. Proc. Inst. Mech. Eng. Part H J. Eng. Med. 1998, 212, 273–280. [Google Scholar] [CrossRef] [PubMed]
- Murugan, R.; Ramakrishna, S. Development of nanocomposites for bone grafting. Compos. Sci. Technol. 2005, 65, 2385–2406. [Google Scholar] [CrossRef]
- Hadjipanayi, E.; Mudera, V.; Brow, R.A. Close dependence of fibroblast proliferation on collagen scaffold matrix stiffness. J. Tissue Eng. Regen. Med. 2009, 13, 512–520. [Google Scholar] [CrossRef]
- Discher, D.E.; Janmey, P.; Wang, Y. Tissue Cells Feel and Respond to the Stiffness of Their Substrate. Mater. Biol. 2005, 310, 1139–1143. [Google Scholar] [CrossRef] [Green Version]
- Guimarães, C.F.; Gasperini, L.; Marques, A.P.; Reis, R.L. The stiffness of living tissues and its implications for tissue engineering. Nat. Rev. Mater. 2020, 5, 351–370. [Google Scholar] [CrossRef]
- Arima, Y.; Iwata, H. Effect of wettability and surface functional groups on protein adsorption and cell adhesion using well-defined mixed self-assembled monolayers. Biomaterials 2007, 28, 3074–3082. [Google Scholar] [CrossRef]
- Morris, V.B.; Nimbalkar, S.; Younesi, M.; McClellan, P.; Akkus, O. Mechanical Properties, Cytocompatibility and Manufacturability of Chitosan:PEGDA Hybrid-Gel Scaffolds by Stereolithography. Ann. Biomed. Eng. 2016, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.; Jha, A.; Harrington, D.A.; Farach-Carson, M. Hyaluronic Acid-Based Hydrogel: From a Natural Polysaccharide to Complex Networks. Soft Matter 2012, 8, 3280–3294. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tyler, B.; Gullotti, D.; Mangraviti, A.; Utsuki, T.; Brem, H. Polylactic acid (PLA) controlled delivery carriers for biomedical applications. Adv. Drug Deliv. Rev. 2016, 107, 163–175. [Google Scholar] [CrossRef] [PubMed]
- Killion, J.A.; Kehoe, S.; Geever, L.M.; Devine, D.M.; Sheehan, E.; Boyd, D.; Higginbotham, C.L. Hydrogel/bioactive glass composites for bone regeneration applications: Synthesis and characterisation. Mater. Sci. Eng. C 2013, 33, 4203–4212. [Google Scholar] [CrossRef]
- Ulery, B.D.; Nair, L.S.; Laurencin, C.T. Biomedical Applications of Biodegradable Polymers. J. Polym. Sci. B Polym. Phys. 2011, 49, 832–864. [Google Scholar] [CrossRef] [Green Version]
- Martinez, P.R.; Goyanes, A.; Basit, A.W.; Gaisford, S. Fabrication of drug-loaded hydrogels with stereolithographic 3D printing. Int. J. Pharm. 2017, 532, 313–317. [Google Scholar] [CrossRef] [Green Version]
- Dhariwala, B.; Hunt, E.; Boland, T. Rapid Prototyping of Tissue-Engineering Constructs, Using. Tissue Eng. 2004, 10, 1316–1322. [Google Scholar] [CrossRef]
- Healy, A.V.; Fuenmayor, E.; Doran, P.; Geever, L.M.; Higginbotham, C.L.; Lyons, J.G. Additive manufacturing of personalized pharmaceutical dosage forms via stereolithography. Pharmaceutics 2019, 11, 645. [Google Scholar] [CrossRef] [Green Version]
- Burke, G.; Barron, V.; Geever, T.; Geever, L.; Devine, D.M.; Higginbotham, C.L. Evaluation of the materials properties, stability and cell response of a range of PEGDMA hydrogels for tissue engineering applications. J. Mech. Behav. Biomed. Mater. 2019, 99, 1–10. [Google Scholar] [CrossRef]
- Burke, G.; Cao, Z.; Devine, D.M.; Major, I. Preparation of biodegradable polyethylene glycol dimethacrylate hydrogels via thiol-ene chemistry. Polymers 2019, 11, 1339. [Google Scholar] [CrossRef] [Green Version]
- Decker, C.; Moussa, K. Photopolymerization of multifunctional monomers in condensed phase. J. Appl. Polym. Sci. 1987, 34, 1603–1618. [Google Scholar] [CrossRef]
- Wu, Y.H.; Park, H.B.; Kai, T.; Freeman, B.D.; Kalika, D.S. Water uptake, transport and structure characterization in poly(ethylene glycol) diacrylate hydrogels. J. Memb. Sci. 2010, 347, 197–208. [Google Scholar] [CrossRef]
- Colthup, N.B.; Daly, L.H.; Wiberley, S.E. Introduction to Infrared and Spectroscopy; Academic Press: London, UK, 1990; ISBN 012182554X. [Google Scholar]
- Coates, J. Interpretation of Infrared Spectra, A Practical Approach Interpretation of Infrared Spectra, A Practical Approach. Encycl. Anal. Chem. 2000, 10815–10837. [Google Scholar] [CrossRef]
- Menczel, J.D.; Prime, R.B. Thermal Analysis of Polymers, 1st ed.; Menczel, J.D., Prime, R.B., Eds.; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2009; ISBN 9780471769170. [Google Scholar]
- Pacios, I.E.; Molina, M.J.; Gómez-Antón, M.R.; Piérola, I.F. Correlation of swelling and crosslinking density with the composition of the reacting mixture employed in radical crosslinking copolymerization. J. Appl. Polym. Sci. 2007, 103, 263–269. [Google Scholar] [CrossRef]
- Omidian, H.; Hashemi, S.A.; Askari, F.; Nafisi, S. Swelling and crosslink density measurements for hydrogels. Iran. J. Polym. Sci. Technol. 1994, 3, 115–119. [Google Scholar]
- Wenzel, R.N. Resistance of solid surfaces to wetting by water. Ind. Eng. Chem. 1936, 28, 988–994. [Google Scholar] [CrossRef]
- Barrett, D.G.; Merkel, T.J.; Luft, J.C.; Yousaf, M.N. One-step syntheses of photocurable polyesters based on a renewable resource. Macromolecules 2010, 43, 9660–9667. [Google Scholar] [CrossRef]
- Zhang, H.; Guo, Y.; Yao, J.; He, M. Epoxidised soybean oil polymer composites reinforced with modified microcrystalline cellulose. J. Exp. Nanosci. 2016, 11, 1213–1226. [Google Scholar] [CrossRef] [Green Version]
- Zhao, J.; Yu, P.; Dong, S. The influence of crosslink density on the failure behavior in amorphous polymers by molecular dynamics simulations. Materials 2016, 9, 234. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Quan, Z.; Cotton, C. Printing direction dependence of mechanical behavior of additively manufactured 3D preforms and composites. Compos. Struct. 2017, 184, 917–923. [Google Scholar] [CrossRef]
- Mueller, J.; Shea, K. The effect of build orientation on the mechanical properties in inkjet 3D printing. In Proceedings of the 26th Annual International Solid Freeform Fabrication Symposium-An Additive Manufacturing Conference, Austin, TX, USA, 10–12 August 2015; pp. 983–992. [Google Scholar]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Polymer | Mean Tg (°C) |
|---|---|
| PEGDMA (UV chamber) chamber) | −23.8 ± 1.4 |
| PEGDMA (SLA) | −24.9 ± 1.5 |
| Polymer | Mean Tg (°C) |
|---|---|
| PEGDMA (UV chamber) chamber) | −42.5 ± 0.7 |
| PEGDMA (SLA) | −41.7 ± 0.3 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Burke, G.; Devine, D.M.; Major, I. Effect of Stereolithography 3D Printing on the Properties of PEGDMA Hydrogels. Polymers 2020, 12, 2015. https://doi.org/10.3390/polym12092015
Burke G, Devine DM, Major I. Effect of Stereolithography 3D Printing on the Properties of PEGDMA Hydrogels. Polymers. 2020; 12(9):2015. https://doi.org/10.3390/polym12092015
Chicago/Turabian StyleBurke, Gavin, Declan M. Devine, and Ian Major. 2020. "Effect of Stereolithography 3D Printing on the Properties of PEGDMA Hydrogels" Polymers 12, no. 9: 2015. https://doi.org/10.3390/polym12092015