Structural Changes of Oak Wood Main Components Caused by Thermal Modification


Abstract
:1. Introduction
2. Materials and Methods
2.1. Samples Preparation
2.2. ATR-FTIR Analysis
2.3. Size-Exclusion Chromatography
3. Results and Discussion
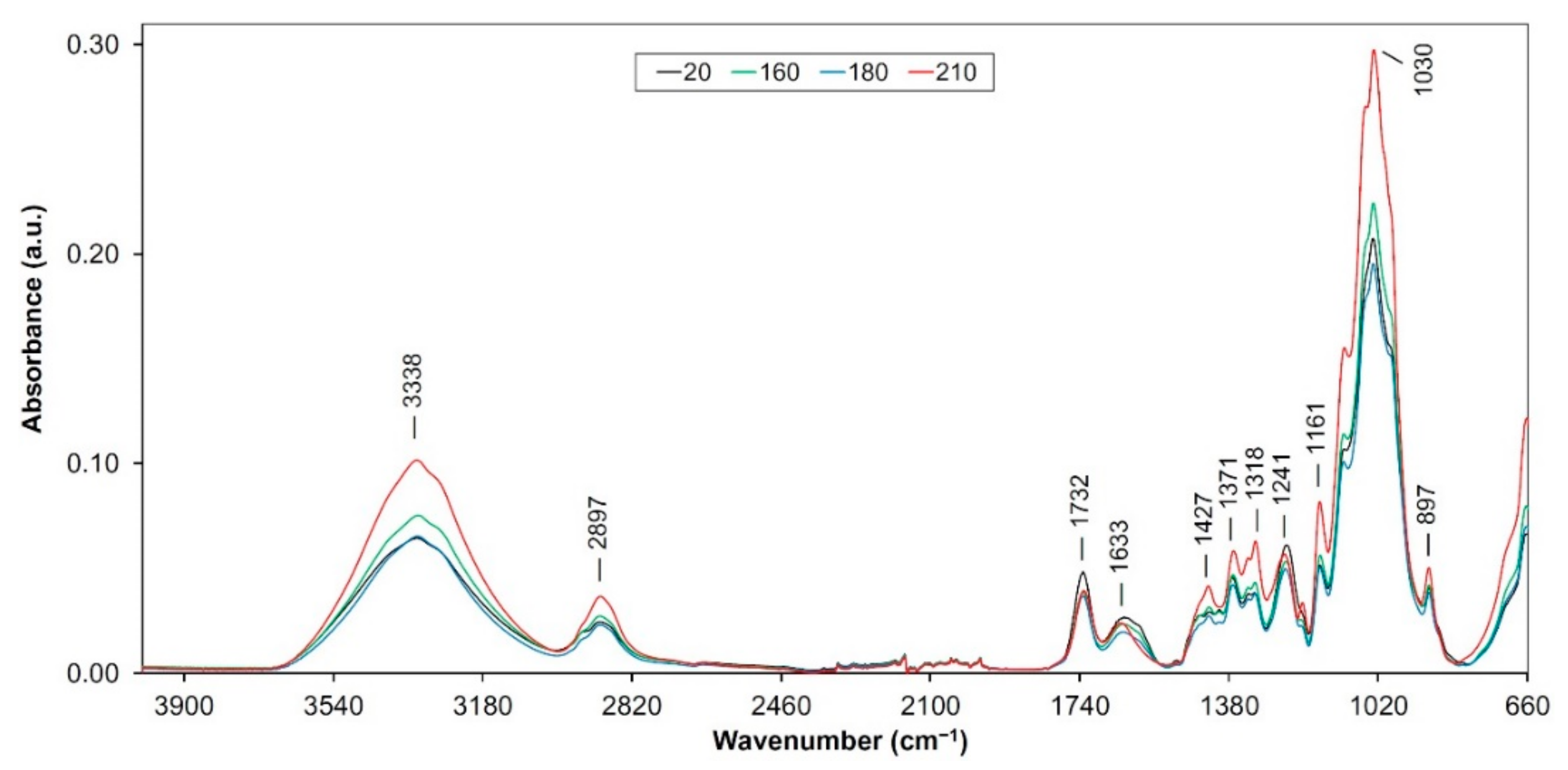
3.1. Changes in the FTIR Spectra
3.1.1. FTIR Spectra of Lignin Isolated from Heat Treated Samples
3.1.2. FTIR Spectra of Holocellulose Complex Isolated from Heat Treated Samples
3.1.3. FTIR Spectra of Cellulose Isolated from Heat Treated Samples
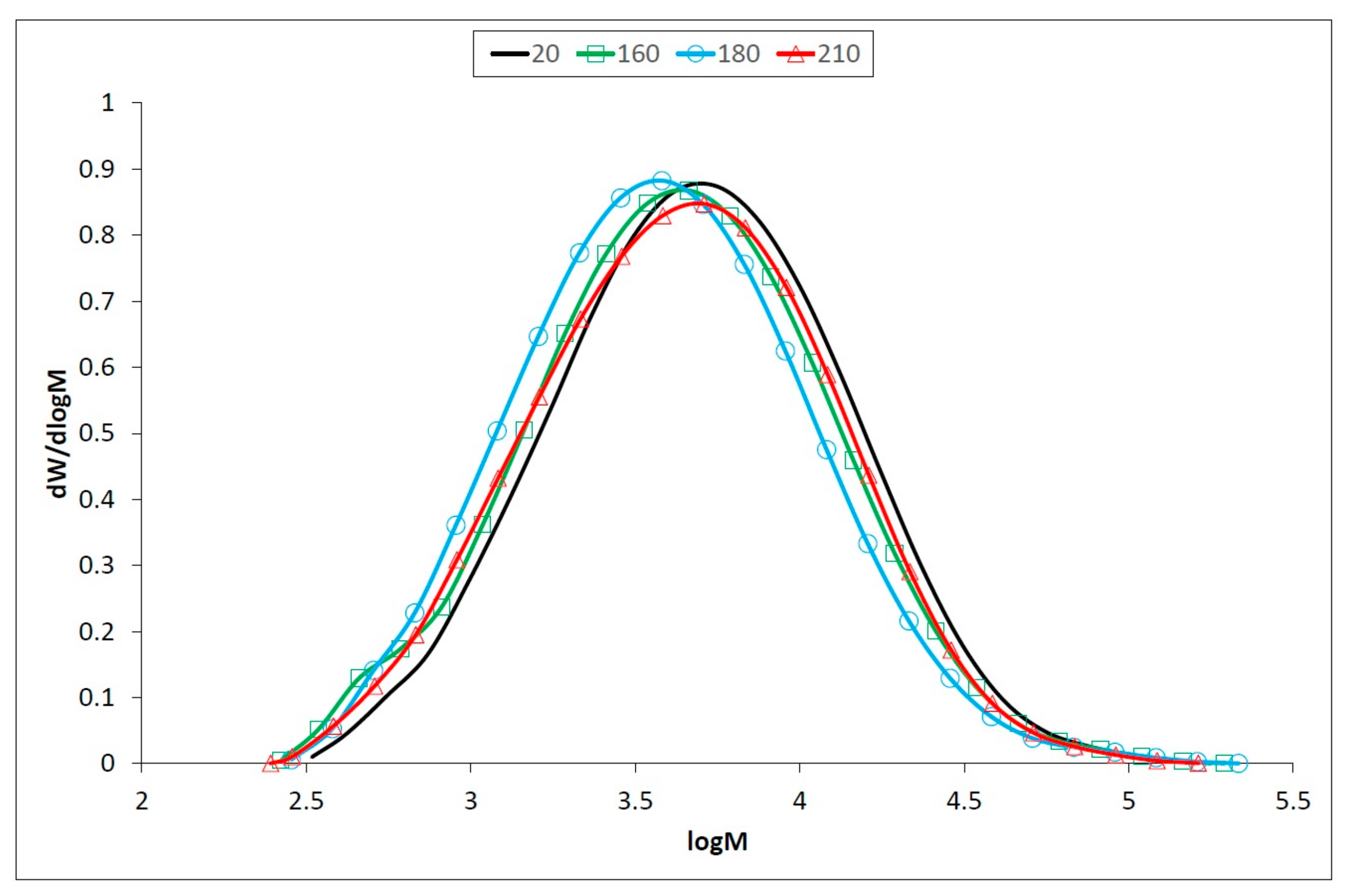
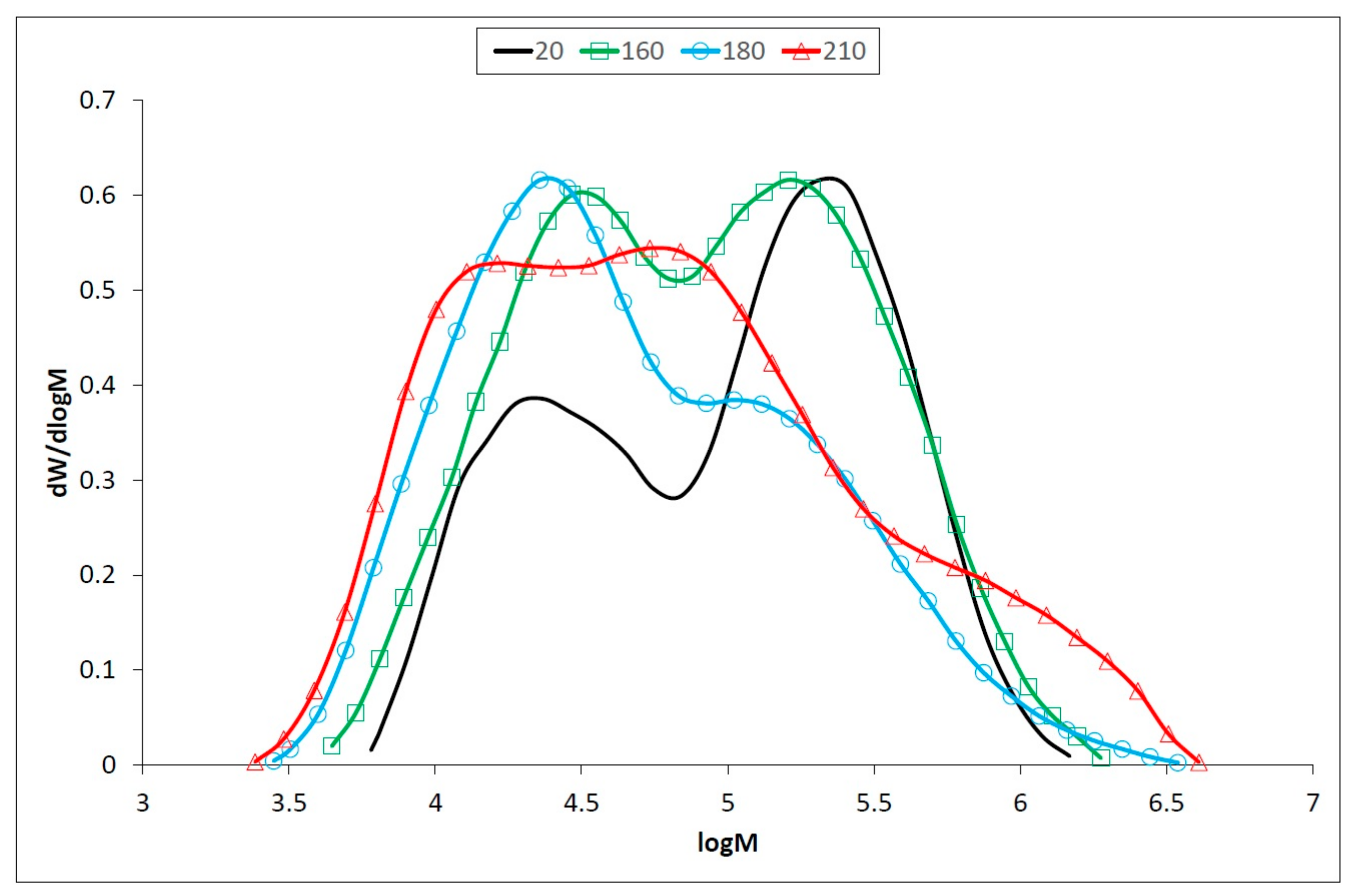
3.2. Changes of Macromolecular Traits in Lignin and Polysaccharides
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Priadi, T.; Hiziroglu, S. Characterization of heat treated wood species. Mater. Des. 2013, 49, 575–582. [Google Scholar] [CrossRef]
- Xu, J.; Zhang, Y.; Shen, Y.; Li, C.; Wang, Y.; Ma, Z.; Sun, W. New perspective on wood thermal modification: Relevance between the evolution of chemical structure and physical-mechanical properties, and online analysis of release of VOCs. Polymers 2019, 11, 1145. [Google Scholar] [CrossRef] [Green Version]
- Okon, K.E.; Lin, F.; Chen, Y.; Huang, B. Effect of silicone oil heat treatment on the chemical composition, cellulose crystalline structure and contact angle of Chinese parasol wood. Carbohydr. Polym. 2017, 164, 179–185. [Google Scholar] [CrossRef]
- Sikora, A.; Kačík, F.; Gaff, M.; Vondrová, V.; Bubeníková, T.; Kubovský, I. Impact of thermal modification on color and chemical changes of spruce and oak wood. J. Wood Sci. 2018, 64, 406–416. [Google Scholar] [CrossRef]
- Gerardin, P. New alternatives for wood preservation based on thermal and chemical modification of wood-a review. Ann. Sci. 2016, 73, 559–570. [Google Scholar] [CrossRef] [Green Version]
- Kubovský, I.; Oberhofnerová, E.; Kačík, F.; Pánek, M. Surface changes of selected hardwoods due to weather conditions. Forests 2018, 9, 557. [Google Scholar] [CrossRef] [Green Version]
- Esteves, B.; Pereira, H. Wood modification by heat treatment: A review. BioResources 2009, 4, 370–404. [Google Scholar]
- Turner, I.; Rousset, P.; Rémond, R.; Perré, P. An experimental and theoretical investigation of the thermal treatment of wood (Fagus sylvatica L.) in the range 200–260 °C. Int. J. Heat Mass Transf. 2010, 53, 715–725. [Google Scholar] [CrossRef]
- Kačíková, D.; Kačík, F.; Čabalová, I.; Ďurkovič, J. Effects of thermal treatment on chemical, mechanical and colour traits in Norway spruce wood. Bioresour. Technol. 2013, 144, 669–674. [Google Scholar] [CrossRef]
- Nuopponen, M.; Vuorinen, T.; Jamsä, S.; Viitaniemi, P. Thermal modifications in softwood studied by FT-IR and UV resonance Raman spectroscopies. J. Wood Chem. Technol. 2004, 24, 13–26. [Google Scholar] [CrossRef]
- Wikberg, H.; Maunu, S.L. Characterization of thermally modified hard and softwoods by 13C CPMAS NMR. Carbohyd. Polym. 2004, 58, 461–466. [Google Scholar] [CrossRef]
- Yildiz, S.; Gümüskaya, E. The effects of thermal modification on crystalline structure of cellulose in soft and hardwood. Build. Environ. 2007, 42, 62–67. [Google Scholar] [CrossRef]
- Kocaefe, D.; Poncsak, S.; Boluk, Y. Effect of thermal treatment on the chemical composition and mechanical properties of birch and aspen. BioResources 2008, 3, 517–537. [Google Scholar] [CrossRef]
- Gaff, M.; Kačík, F.; Sandberg, D.; Babiak, M.; Turčáni, M.; Niemz, P.; Hanzlík, P. The effect of chemical changes during thermal modification of European oak and Norway spruce on elasticity properties. Compos. Struct. 2019, 220, 529–538. [Google Scholar] [CrossRef]
- Gaff, M.; Babiak, M.; Kačík, F.; Sandberg, D.; Turčáni, M.; Hanzlík, P.; Vondrová, V. Plasticity properties of thermally modified timber in bending—The effect of chemical changes during modification of European oak and Norway spruce. Compos. Part B Eng. 2019, 165, 613–625. [Google Scholar] [CrossRef]
- Kačík, F.; Luptáková, J.; Šmíra, P.; Nasswettrová, A.; Kačíková, D.; Vacek, V. Chemical alterations of pine wood lignin during heat sterilization. BioResources 2016, 11, 3442–3452. [Google Scholar] [CrossRef] [Green Version]
- Kačík, F.; Kačíková, D.; Jablonský, M.; Katuščák, S. Cellulose degradation in newsprint paper ageing. Polym. Degrad. Stabil. 2009, 94, 1509–1514. [Google Scholar] [CrossRef]
- Faix, O. Classification of lignins from different botanical origins by FTIR spectroscopy. Holzforschung 1991, 45, 21–27. [Google Scholar] [CrossRef]
- Cheng, S.; Huang, A.; Wang, S.; Zhang, Q. Effect of Different Heat Treatment Temperatures on the Chemical Composition and Structure of Chinese Fir Wood. BioResources 2016, 11, 4006–4016. [Google Scholar] [CrossRef] [Green Version]
- Shen, D.K.; Gu, S. The mechanism for thermal decomposition of cellulose and its main products. Bioresour. Technol. 2009, 100, 6496–6504. [Google Scholar] [CrossRef]
- Esteves, B.; Marques, A.V.; Domingos, I.; Pereira, H. Chemical changes of heat treated pine and eucalypt wood monitored by FTIR. Maderas Cienc. Tecnol. 2013, 15, 245–258. [Google Scholar] [CrossRef] [Green Version]
- Mattos, B.D.; Lourençon, T.V.; Serrano, L.; Labidi, J.; Gatto, D.A. Chemical modification of fast-growing eucalyptus wood. Wood Sci. Technol. 2015, 2, 273–288. [Google Scholar] [CrossRef]
- Missio, A.L.; Mattos, B.D.; de Cademartori, P.H.; Pertuzzatti, A.; Conte, B.; Gatto, D.A. Thermochemical and physical properties of two fast-growing eucalypt woods subjected to two-step freeze-heat treatments. Acta 2015, 615, 15–22. [Google Scholar] [CrossRef]
- Inari, G.N.; Petrissans, M.; Gerardin, P. Chemical reactivity of heat-treated wood. Wood Sci. Technol. 2007, 41, 157–168. [Google Scholar] [CrossRef]
- Spiridon, I.; Teaca, C.A.; Bodȋrlǎu, R. Structural changes evidenced by FTIR spectroscopy in cellulosic materials after pretreatment with ionic liquid and enzymatic hydrolysis. BioResources 2011, 6, 400–413. [Google Scholar]
- Ates, S.; Akyildiz, M.H.; Ozdemir, H. Effects of heat treatment on calabrian pine (Pinus brutia Ten.) wood. BioResources 2009, 4, 1032–1043. [Google Scholar] [CrossRef]
- Li, J.; Li, B.; Zhang, X. Comparative studies of heat degradation between larch lignin and Manchurian ash lignin. Polym. Degrad. Stabil. 2002, 78, 279–285. [Google Scholar] [CrossRef]
- Kačík, F.; Kačíková, D.; Bubeníková, T. Spruce wood lignin alteration after infrared heating at different wood moistures. Cell. Chem. Technol. 2006, 40, 643–648. [Google Scholar]
- Vartanian, E.; Barres, O.; Roque, C. FTIR spectroscopy of woods: A new approach to study the weathering of the carving face of a sculpture. Spectrochim. Acta A 2015, 136, 1255–1259. [Google Scholar] [CrossRef]
- Özgenç, Ö.; Durmaz, S.; Boyaci, I.H.; Eksi-Kocak, H. Determination of chemical changes in heat-treated wood using ATR-FTIR and FT Raman spectrometry. Spectrochim. Acta A 2017, 171, 395–400. [Google Scholar] [CrossRef]
- Michell, A.; Higgins, H. Infrared Spectroscopy in Australian Forest Products Research; CSIRO Forestry and Forest Products: Melbourne, Australia, 2002; p. 60. [Google Scholar]
- González-Peña, M.M.; Curling, S.F.; Hale, M.D. On the effect of heat on the chemical composition and dimensions of thermally-modified wood. Polym. Degrad. Stabil. 2009, 94, 2184–2193. [Google Scholar] [CrossRef]
- Li, M.Y.; Cheng, S.C.; Li, D.; Wang, S.N.; Huang, A.M.; Sun, S.Q. Structural characterization of steam-heat treated Tectona grandis wood analyzed by FT-IR and 2D-IR correlation spectroscopy. Chin. Chem. Lett. 2015, 26, 221–225. [Google Scholar] [CrossRef]
- Kubovský, I.; Kačík, F. Colour and chemical changes of the lime wood surface due to CO2 laser thermal modification. Appl. Surf. Sci. 2014, 321, 261–267. [Google Scholar] [CrossRef]
- Wang, S.; Ru, B.; Dai, G.; Sun, W.; Qiu, K.; Zhou, J. Pyrolysis mechanism study of minimally damaged hemicellulose polymers isolated from agricultural waste straw samples. Bioresour. Technol. 2015, 190, 211–218. [Google Scholar] [CrossRef] [PubMed]
- Colom, X.; Carrillo, F.; Nogués, F.; Garriga, P. Structural analysis of photodegraded wood by means of FTIR spectroscopy. Polym. Degrad. Stabil. 2003, 80, 543–549. [Google Scholar] [CrossRef]
- Papp, G.; Barta, E.; Preklet, E.; Tolvaj, L.; Berkesi, O.; Nagy, T.; Szatmari, S. Changes in DRIFT spectra of wood irradiated by UV laser as a function of energy. J. Photochem. Photobio. A 2005, 173, 137–142. [Google Scholar] [CrossRef]
- Bourgois, J.; Guyonnet, R. Characterization and analysis of torrefied wood. Wood Sci. Technol. 1988, 22, 143–155. [Google Scholar] [CrossRef]
- Yang, H.; Rong, Y.; Chen, H.; Dong, H.L.; Zheng, C. Characteristics of hemicellulose, cellulose and lignin pyrolysis. Fuel 2007, 86, 1781–1788. [Google Scholar] [CrossRef]
- Lin, B.J.; Colin, B.; Chen, W.H.; Pétrissans, A.; Rousset, P.; Pétrissans, M. Thermal degradation and compositional changes of wood treated in a semi-industrial scale reactor in vacuum. J. Anal. Appl. Pyrol. 2018, 130, 8–18. [Google Scholar] [CrossRef]
- Collier, W.E.; Schultz, T.P.; Kalasinsky, V.F. Infrared Study of Lignin: Reexamination of Aryl-Alkyl Ether C-O Stretching Peak Assignments. Holzforschung 1992, 46, 523–528. [Google Scholar] [CrossRef]
- Hosseinaei, O.; Wang, S.; Enayati, A.A.; Rials, T.G. Effects of hemicellulose extraction on properties of wood flour and wood-plastic composites. Compos. Part A Appl. Sci. 2012, 43, 686–694. [Google Scholar] [CrossRef]
- Popescu, M.C.; Froidevaux, J.; Navi, P.; Popescu, C.M. Structural modifications of Tilia cordata wood during heat treatment investigated by FT-IR and 2D IR correlation spectroscopy. J. Mol. Struct. 2013, 1033, 176–186. [Google Scholar] [CrossRef]
- Miklečić, J.; Jirouš-Rajković, V.; Antonović, A.; Španić, N. Discolouration of thermally modified wood during simulated indoor sunlight exposure. BioResources 2011, 6, 434–446. [Google Scholar] [CrossRef]
- Faix, O. Fourier transform infrared spectroscopy. In Methods in Lignin Chemistry; Lin, S.Y., Dence, C.W., Eds.; Springer: Berlin/Heidelberg, Germany, 1992; Chapter 4.1; pp. 83–109. [Google Scholar] [CrossRef]
- Windeisen, E.; Strobel, C.; Wegener, G. Chemical changes during the production of thermo-treated beech wood. Wood Sci. Technol. 2007, 41, 523–536. [Google Scholar] [CrossRef]
- Peng, W.; Wang, L.; Ohkoshi, M.; Zhang, M. Separation of hemicelluloses from Eucalyptus species: Investigating the residue after alkaline treatment. Cell. Chem. Technol. 2015, 49, 756–764. [Google Scholar]
- Arif, S.; Kautek, W. Laser cleaning of particulates from paper: Comparison between sized ground wood cellulose and pure cellulose. Appl. Surf. Sci. 2013, 276, 53–61. [Google Scholar] [CrossRef]
- Rodrigues, J.; Faix, O.; Pereira, H. Determination of lignin content of Eucalyptus globulus wood using FTIR spectroscopy. Holzforschung 1998, 52, 46–50. [Google Scholar] [CrossRef]
- Fengel, D.; Ludwig, M. Möglichkeiten und Grenzen der FTIR-Spektroskopie bei der Charakterisierung von Cellulose. Teil 1. Vergleich von verschiedenen Cellulosefasern und Bakterien-Cellulose. Das Pap. 1991, 45, 45–51. [Google Scholar]
- Kuo, M.L.; McClelland, J.F.; Luo, S.; Chien, P.L.; Walker, R.D.; Hse, C.Y. Applications of infrared photoacoustic spectroscopy for wood samples. Wood Fiber Sci. 1988, 20, 132–145. [Google Scholar]
- Åkerholm, M.; Hinterstoisser, B.; Salmén, L. Characterization of the crystalline structure of cellulose using static and dynamic FT-IR spectroscopy. Carbohydr. Res. 2004, 339, 569–578. [Google Scholar] [CrossRef]
- Alén, R.; Kotilainen, R.; Zaman, A. Thermochemical behavior of Norway spruce (Picea abies) at 180–225 °C. Wood Sci. Technol. 2002, 36, 163–171. [Google Scholar] [CrossRef]
- Esteves, B.; Domingos, I.; Pereira, H. Pine wood modification by heat treatment in air. BioResources 2008, 3, 142–154. [Google Scholar] [CrossRef]
- Weiland, J.J.; Guyonnet, R. Study of chemical modifications and fungi degradation of thermally modified wood using DRIFT spectroscopy. Holz Roh Werkst. 2003, 61, 216–220. [Google Scholar] [CrossRef]
- Faravelli, T.; Frassoldati, A.; Migliavacca, G.; Ranzi, E. Detailed kinetic modeling of the thermal degradation of lignins. Biomass Bioenerg. 2010, 34, 290–301. [Google Scholar] [CrossRef]
- Zhang, Y.; Ma, Z.; Zhang, Q.; Wang, J.; Ma, Q.; Yang, Y.; Luo, X.; Zhang, W. Comparison of the physicochemical characteristics of bio-char pyrolyzed from moso bamboo and rice husk with different pyrolysis temperatures. BioResources 2017, 12, 4652–4669. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.Y.; Hwang, H.; Oh, S.; Kim, Y.S.; Kim, U.J.; Choi, J.W. Investigation of structural modification and thermal characteristics of lignin after heat treatment. Int. J. Biol. Macromol. 2014, 66, 57–65. [Google Scholar] [CrossRef]
- Kato, K.L.; Cameron, R.E. A review of the relationship between thermally accelerated ageing of paper and hornification. Cellulose 1999, 6, 23–40. [Google Scholar] [CrossRef]
- Kačík, F.; Šmíra, P.; Kačíková, D.; Veľková, V.; Nasswettrová, A.; Vacek, V. Chemical alterations of pine wood saccharides during heat sterilisation. Carbohyd. Polym. 2015, 117, 681–686. [Google Scholar] [CrossRef]
- Hrčka, R.; Kučerová, V.; Hýrošová, T. Correlation between Oak Wood Properties. BioResources 2018, 13, 8885–8898. [Google Scholar] [CrossRef]
- Prinks, M.J.; Ptasinski, K.J.; Jansen, F.J.J.G. Torrefection of wood, part 2. Analysis of products. J. Anal. Appl. Pyrol. 2006, 77, 35–40. [Google Scholar] [CrossRef]
- Candelier, K.; Thevenon, M.F.; Petrissans, A.; Dumarcay, S.; Gerardin, P.; Petrissans, M. Control of wood thermal treatment and its effects on decay resistance: A review. Ann. Sci. 2016, 73, 571. [Google Scholar] [CrossRef] [Green Version]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| T (°C) | Mn | Mw | Mz | Mz+1 | PDI |
|---|---|---|---|---|---|
| 20 | 3474 (19) | 9216 (138) | 23,004 (672) | 47,401 (1786) | 2.65 (0.03) |
| 160 | 2987 (34) | 8477 (22) | 23,838 (97) | 54,001 (310) | 2.84 (0.03) |
| 180 | 2736 (23) | 7650 (24) | 24,899 (644) | 63,155 (1626) | 2.80 (0.03) |
| 210 | 3021 (46) | 8435 (20) | 21,517 (268) | 44,751 (1075) | 2.79 (0.05) |
| T (°C) | Mn | Mw | Mz | Mz+1 | PDI | DP |
|---|---|---|---|---|---|---|
| 20 | 51,097 (542) | 212,995 (8405) | 461,299 (33,735) | 699,959 (65,649) | 4.17 (0.18) | 1315 (52) |
| 160 | 44,400 (577) | 186,382 (3793) | 487,413 (13,473) | 817,872 (33,397) | 4.20 (0.09) | 1151 (23) |
| 180 | 27,524 (353) | 146,648 (8298) | 640,517 (33,698) | 1,339,376 (61,086) | 5.33 (0.25) | 905 (51) |
| 210 | 29,042 (327) | 275,447 (12,292) | 1,223,920 (28,533) | 1,929,683 (24,264) | 9.48 (0.35) | 1700 (76) |
| T (°C) | Mn | Mw | Mz | Mz+1 | PDI | DP |
|---|---|---|---|---|---|---|
| 20 | 39,375 (1165) | 385,896 (2376) | 1,150,316 (14,034) | 1,808,690 (26,230) | 9.81 (0.31) | 2382 (15) |
| 160 | 35,634 (438) | 384,908 (607) | 1,130,145 (4320) | 1,758,540 (18,341) | 10.80 (0.14) | 2376 (4) |
| 180 | 30,154 (226) | 355,894 (1091) | 1,190,572 (4984) | 1,907,652 (8892) | 11.80 (0.10) | 2197 (7) |
| 210 | 33,560 (232) | 306,850 (2385) | 1,044,946 (10,935) | 1,728,619 (19,617) | 9.14 (0.13) | 1894 (15) |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kubovský, I.; Kačíková, D.; Kačík, F. Structural Changes of Oak Wood Main Components Caused by Thermal Modification. Polymers 2020, 12, 485. https://doi.org/10.3390/polym12020485
Kubovský I, Kačíková D, Kačík F. Structural Changes of Oak Wood Main Components Caused by Thermal Modification. Polymers. 2020; 12(2):485. https://doi.org/10.3390/polym12020485
Chicago/Turabian StyleKubovský, Ivan, Danica Kačíková, and František Kačík. 2020. "Structural Changes of Oak Wood Main Components Caused by Thermal Modification" Polymers 12, no. 2: 485. https://doi.org/10.3390/polym12020485