Study on the Anti-Biodegradation Property of Tunicate Cellulose


Abstract
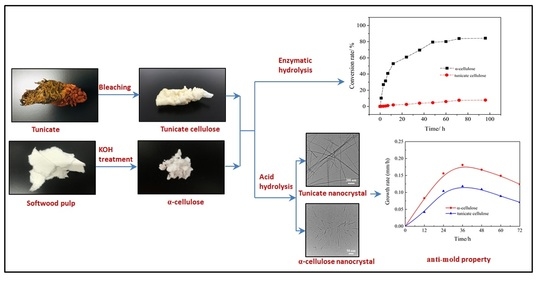
:
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Methods
2.2.1. Preparation of Tunicate Cellulose
2.2.2. Preparation of Amorphous Cellulose
2.2.3. Preparation of Softwood α-Cellulose
2.2.4. Enzymatic Hydrolysis of Cellulose Samples
2.2.5. Characterization
XRD
Degree of Polymerization
FTIR Analysis
2.2.6. Anti-Mold Experiment
Preparation of Cellulose Nanocrystals
TEM Analysis of Prepared Softwood Cellulose Nanocrystal
Anti-Mold Effect of Cellulose Nanocrystals
Growth Rate Analysis
2.3. Error Analysis
3. Results and Discussion
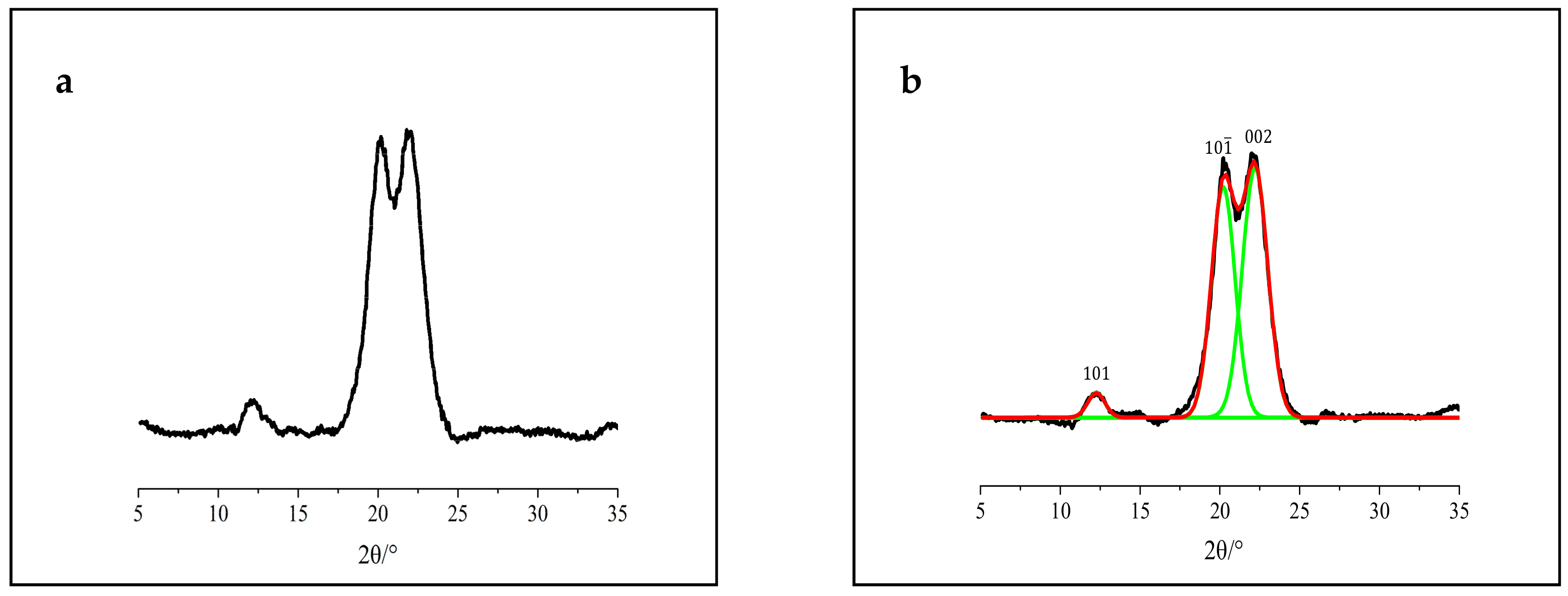
3.1. XRD Analysis
3.2. FTIR Analysis
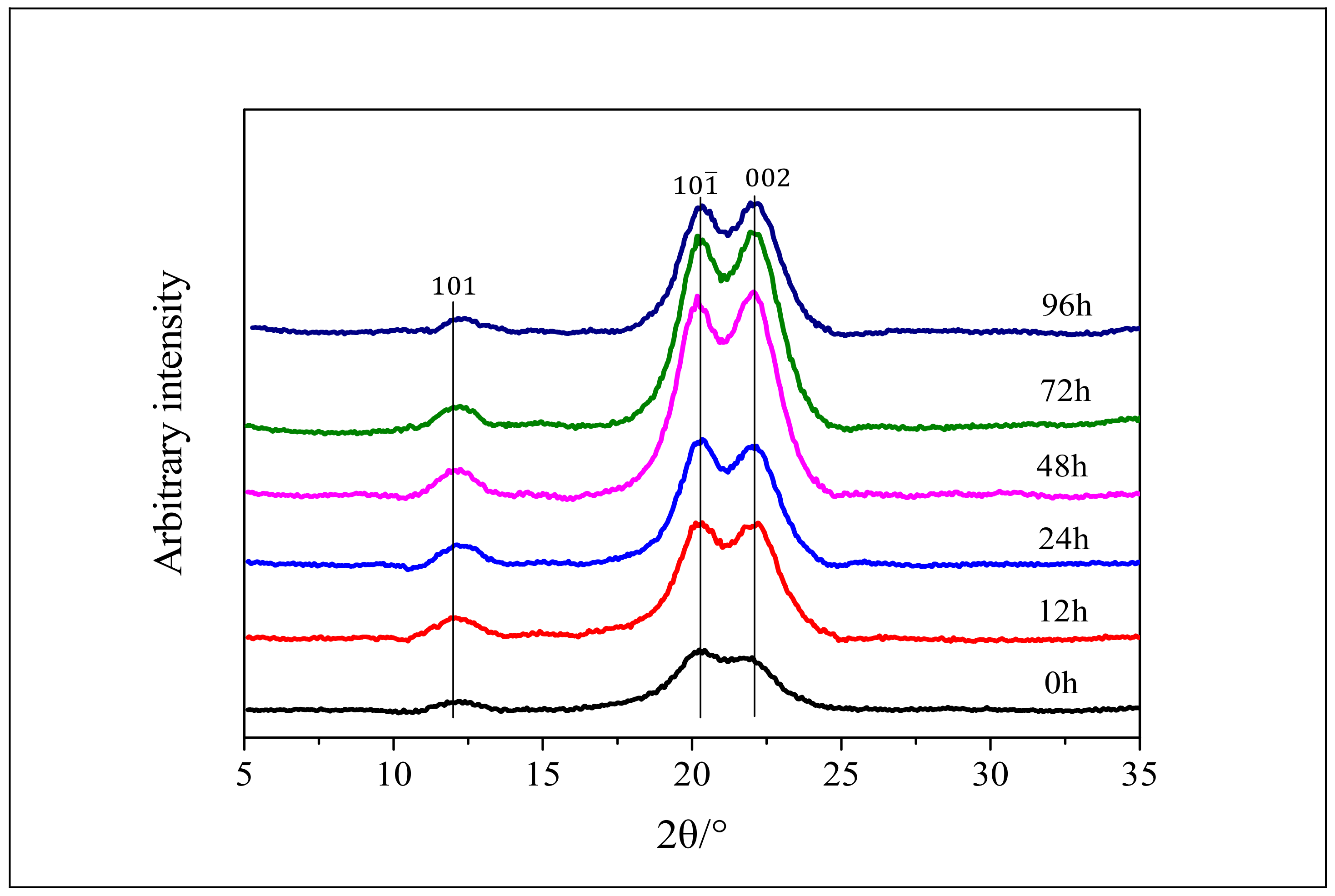
3.3. XRD Analysis of Cellulose after Enzymatic Hydrolysis
3.4. Effects of Enzymatic Hydrolysis on CrI and Average Crystal Size of Cellulose
3.5. Degree of Polymerization
3.6. Cellulose Conversion Rate in Enzymatic Hydrolysis
3.7. TEM Analysis of Cellulose Nanocrystals
3.8. Anti-Mold Effects of Tunicate and α-Cellulose Nanocrystals
3.9. Measurement of Mold Growth Rate in the Medium Containing Various Amounts of Tunicate Nanocrystals
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Rojas, O.J. Cellulose Chemistry and Properties: Fibers, Nanocelluloses and Advanced Materials; Springer: Berlin/Heidelberg, Germany, 2016; Volume 271. [Google Scholar]
- Zhao, Y.; Li, J. Excellent chemical and material cellulose from tunicates: Diversity in cellulose production yield and chemical and morphological structures from different tunicate species. Cellulose 2014, 21, 3427–3441. [Google Scholar] [CrossRef]
- Habibi, Y.; Lucia, L.A.; Rojas, O.J. Cellulose nanocrystals: Chemistry, self-assembly, and applications. Chem. Rev. 2010, 110, 3479–3500. [Google Scholar] [CrossRef]
- Schroers, M.; Kokil, A.; Weder, C. Solid polymer electrolytes based on nanocomposites of ethylene oxide–epichlorohydrin copolymers and cellulose whiskers. J. Appl. Polym. Sci. 2004, 93, 2883–2888. [Google Scholar] [CrossRef]
- Mendez, J.D.; Weder, C. Synthesis, electrical properties, and nanocomposites of poly (3, 4-ethylenedioxythiophene) nanorods. Polym. Chem. 2010, 1, 1237–1244. [Google Scholar] [CrossRef] [Green Version]
- Van den Berg, O.; Schroeter, M.; Capadona, J.R.; Weder, C. Nanocomposites based on cellulose whiskers and (semi) conducting conjugated polymers. J. Mater. Chem. 2007, 17, 2746–2753. [Google Scholar] [CrossRef]
- Podsiadlo, P.; Sui, L.; Elkasabi, Y.; Burgardt, P.; Lee, J.; Miryala, A.; Kusumaatmaja, W.; Carman, M.R.; Shtein, M.; Kieffer, J. Layer-by-layer assembled films of cellulose nanowires with antireflective properties. Langmuir 2007, 23, 7901–7906. [Google Scholar] [CrossRef]
- Zhu, G.; Xu, H.; Dufresne, A.; Lin, N. High-adsorption, self-extinguishing, thermal, and acoustic-resistance aerogels based on organic and inorganic waste valorization from cellulose nanocrystals and red mud. ACS Sustain. Chem. Eng. 2018, 6, 7168–7180. [Google Scholar] [CrossRef]
- Biyani, M.V.; Foster, E.J.; Weder, C. Light-healable supramolecular nanocomposites based on modified cellulose nanocrystals. ACS Macro Lett. 2013, 2, 236–240. [Google Scholar] [CrossRef]
- Fox, J.; Wie, J.J.; Greenland, B.W.; Burattini, S.; Hayes, W.; Colquhoun, H.M.; Mackay, M.E.; Rowan, S.J. High-strength, healable, supramolecular polymer nanocomposites. J. Am. Chem. Soc. 2012, 134, 5362–5368. [Google Scholar] [CrossRef] [Green Version]
- Capadona, J.R.; Shanmuganathan, K.; Tyler, D.J.; Rowan, S.J.; Weder, C. Stimuli-responsive polymer nanocomposites inspired by the sea cucumber dermis. Science 2008, 319, 1370–1374. [Google Scholar] [CrossRef]
- Kim, D.S.; Jung, S.-M.; Yoon, G.H.; Lee, H.C.; Shin, H.S. Development of a complex bone tissue culture system based on cellulose nanowhisker mechanical strain. Colloids Surf. B Biointerfaces 2014, 123, 838–844. [Google Scholar] [CrossRef] [PubMed]
- American Chemical Society. Contaminants in Our Water: Identification and Remediation Methods; ACS Publications: Washington, DC, USA, 2020. [Google Scholar]
- Mahfoudhi, N.; Boufi, S. Nanocellulose as a novel nanostructured adsorbent for environmental remediation: A review. Cellulose 2017, 24, 1171–1197. [Google Scholar] [CrossRef]
- Thomas, B.; Raj, M.C.; Joy, J.; Moores, A.; Drisko, G.L.; Sanchez, C.m. Nanocellulose, a versatile green platform: From biosources to materials and their applications. Chem. Rev. 2018, 118, 11575–11625. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.; Yu, H.; Lee, S.-Y.; Wei, T.; Li, J.; Fan, Z. Nanocellulose: A promising nanomaterial for advanced electrochemical energy storage. Chem. Soc. Rev. 2018, 47, 2837–2872. [Google Scholar] [CrossRef]
- Wang, L.; Zuo, X.; Raut, A.; Isseroff, R.; Xue, Y.; Zhou, Y.; Sandhu, B.; Schein, T.; Zeliznyak, T.; Sharma, P. Operation of proton exchange membrane (PEM) fuel cells using natural cellulose fiber membranes. Sustain. Energy Fuels 2019, 3, 2725–2732. [Google Scholar] [CrossRef]
- Klemm, D.; Cranston, E.D.; Fischer, D.; Gama, M.; Kedzior, S.A.; Kralisch, D.; Kramer, F.; Kondo, T.; Lindström, T.; Nietzsche, S. Nanocellulose as a natural source for groundbreaking applications in materials science: Today’s state. Mater. Today 2018, 21, 720–748. [Google Scholar] [CrossRef] [Green Version]
- Christov, L.; Akhtar, M.; Prior, B. The potential of biosulfite pulping in dissolving pulp production. Enzym. Microb. Technol. 1998, 23, 70–74. [Google Scholar] [CrossRef]
- Adney, B.; Baker, J. Measurement of Cellulase Activities: Laboratory Analytical Procedure (LAP); Technical Report; National Renewable Energy Laboratory (NREL): Golden, CO, USA, 2008.
- Ouyang, J.; Dong, Z.; Song, X.; Lee, X.; Chen, M.; Yong, Q. Improved enzymatic hydrolysis of microcrystalline cellulose (Avicel PH101) by polyethylene glycol addition. Bioresour. Technol. 2010, 101, 6685–6691. [Google Scholar] [CrossRef]
- Park, S.; Baker, J.O.; Himmel, M.E.; Parilla, P.A.; Johnson, D.K. Cellulose crystallinity index: Measurement techniques and their impact on interpreting cellulase performance. Biotechnol. Biofuels 2010, 3, 10. [Google Scholar] [CrossRef] [Green Version]
- Segal, L.; Creely, J.; Martin, A., Jr.; Conrad, C. An empirical method for estimating the degree of crystallinity of native cellulose using the X-ray diffractometer. Text. Res. J. 1959, 29, 786–794. [Google Scholar] [CrossRef]
- Heinze, T.; Liebert, T. Unconventional methods in cellulose functionalization. Prog. Polym. Sci. 2001, 26, 1689–1762. [Google Scholar] [CrossRef]
- Hubbell, C.A.; Ragauskas, A.J. Effect of acid-chlorite delignification on cellulose degree of polymerization. Bioresour. Technol. 2010, 101, 7410–7415. [Google Scholar] [CrossRef] [PubMed]
- Cohen, R.; Jensen, K.A.; Houtman, C.J.; Hammel, K.E. Significant levels of extracellular reactive oxygen species produced by brown rot basidiomycetes on cellulose. Febs Lett. 2002, 531, 483–488. [Google Scholar] [CrossRef] [Green Version]
- Tang, Y.; Yang, S.; Zhang, N.; Zhang, J. Preparation and characterization of nanocrystalline cellulose via low-intensity ultrasonic-assisted sulfuric acid hydrolysis. Cellulose 2014, 21, 335–346. [Google Scholar] [CrossRef]
- Boriová, K.; Čerňanský, S.; Matúš, P.; Bujdoš, M.; Šimonovičová, A.; Urík, M. Removal of aluminium from aqueous solution by four wild-type strains of Aspergillus niger. Bioprocess Biosyst. Eng. 2019, 42, 291–296. [Google Scholar] [CrossRef] [PubMed]
- Zhao, P.; Xia, W.; Lei, C.; Omer, S.H.; Zhang, X.; Zhuang, Y.; Chen, S. Isolation, Identification and Physiological Activity of Endophytes from the Roots of Vitis vinifera. Nanosci. Nanotechnol. Lett. 2016, 8, 532–538. [Google Scholar] [CrossRef]
- Siede, W. A “Hole Punched Plate” method for easy generation and harvesting of microconidia in the dermatophyte Trichophyton rubrum. Heliyon 2018, 4, e00676. [Google Scholar] [CrossRef]
- Haafiz, M.M.; Eichhorn, S.; Hassan, A.; Jawaid, M. Isolation and characterization of microcrystalline cellulose from oil palm biomass residue. Carbohydr. Polym. 2013, 93, 628–634. [Google Scholar] [CrossRef]
- Abraham, E.; Deepa, B.; Pothan, L.; Jacob, M.; Thomas, S.; Cvelbar, U.; Anandjiwala, R. Extraction of nanocellulose fibrils from lignocellulosic fibres: A novel approach. Carbohydr. Polym. 2011, 86, 1468–1475. [Google Scholar] [CrossRef]
- Kale, R.D.; Bansal, P.S.; Gorade, V.G. Extraction of microcrystalline cellulose from cotton sliver and its comparison with commercial microcrystalline cellulose. J. Polym. Environ. 2018, 26, 355–364. [Google Scholar] [CrossRef]
- Zhao, Y.; Moser, C.; Lindström, M.E.; Henriksson, G.; Li, J. Cellulose nanofibers from softwood, hardwood, and tunicate: Preparation–structure–film performance interrelation. ACS Appl. Mater. Interfaces 2017, 9, 13508–13519. [Google Scholar] [CrossRef] [PubMed]
- Trilokesh, C.; Uppuluri, K.B. Isolation and characterization of cellulose nanocrystals from jackfruit peel. Sci. Rep. 2019, 9, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Ibrahim, N.A.; Azraaie, N.; Abidin, Z.; Mohd, N.A.; Mamat Razali, N.A.; Abdul Aziz, F.; Zakaria, S. Preparation and characterization of alpha cellulose of pineapple (Ananas comosus) leaf fibres (PALF). In Advanced Materials Research; NREL: Golden, CO, USA, 2014; pp. 147–150. [Google Scholar]
- Rivai, H.; Hamdani, A.S.; Ramdani, R.; Lalfari, R.S.; Andayani, R.; Armin, F.; Djamaan, A. Production and Characterization of Alpha Cellulose Derived From Rice Straw (Oryza sativa L.). Int. J. Pharm. Sci. Rev. Res. 2018, 52, 45–48. [Google Scholar]
- Galiwango, E.; Rahman, N.S.A.; Al-Marzouqi, A.H.; Abu-Omar, M.M.; Khaleel, A.A. Isolation and characterization of cellulose and α-cellulose from date palm biomass waste. Heliyon 2019, 5, e02937. [Google Scholar] [CrossRef] [Green Version]
- Ju, X.; Bowden, M.; Brown, E.E.; Zhang, X. An improved X-ray diffraction method for cellulose crystallinity measurement. Carbohydr. Polym. 2015, 123, 476–481. [Google Scholar] [CrossRef] [Green Version]
- Ciolacu, D.; Ciolacu, F.; Popa, V.I. Amorphous cellulose—Structure and characterization. Cellul. Chem. Technol. 2011, 45, 13. [Google Scholar]
- Sugiyama, J.; Persson, J.; Chanzy, H. Combined infrared and electron diffraction study of the polymorphism of native celluloses. Macromolecules 1991, 24, 2461–2466. [Google Scholar] [CrossRef]
- Rosa, M.; Medeiros, E.; Malmonge, J.; Gregorski, K.; Wood, D.; Mattoso, L.; Glenn, G.; Orts, W.; Imam, S. Cellulose nanowhiskers from coconut husk fibers: Effect of preparation conditions on their thermal and morphological behavior. Carbohydr. Polym. 2010, 81, 83–92. [Google Scholar] [CrossRef]
- Popescu, M.-C.; Popescu, C.-M.; Lisa, G.; Sakata, Y. Evaluation of morphological and chemical aspects of different wood species by spectroscopy and thermal methods. J. Mol. Struct. 2011, 988, 65–72. [Google Scholar] [CrossRef]
- Poletto, M.; Pistor, V.; Zeni, M.; Zattera, A.J. Crystalline properties and decomposition kinetics of cellulose fibers in wood pulp obtained by two pulping processes. Polym. Degrad. Stab. 2011, 96, 679–685. [Google Scholar] [CrossRef]
- Zhao, Y.; Zhang, Y.; Lindström, M.E.; Li, J. Tunicate cellulose nanocrystals: Preparation, neat films and nanocomposite films with glucomannans. Carbohydr. Polym. 2015, 117, 286–296. [Google Scholar] [CrossRef] [PubMed]
- Szymanska-Chargot, M.; Zdunek, A. Use of FT-IR spectra and PCA to the bulk characterization of cell wall residues of fruits and vegetables along a fraction process. Food Biophys. 2013, 8, 29–42. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fengel, D.; Wegener, G. Wood: Chemistry, ultrastructure. Reactions 1984, 613, 1960–1982. [Google Scholar]
- Teeri, T.T. Crystalline cellulose degradation: New insight into the function of cellobiohydrolases. Trends Biotechnol. 1997, 15, 160–167. [Google Scholar] [CrossRef]
- Fan, L.; Gharpuray, M.; Lee, Y.H. Evaluation of pretreatments for enzymatic conversion of agricultural residues. In Proceedings of the Symposium on Biotechnology in Energy Production and Conservation, Gatlinburg, TN, USA, 12 May 1981. [Google Scholar]
- Li, Q.; Qi, W.; Su, R.; He, Z. Enhanced enzymatic hydrolysis and changes in structure of cellulose regenerated with ionic liquids. Cellul. Chem. Technol. 2017, 51, 593–600. [Google Scholar]
- Sasaki, T.; Tanaka, T.; Nanbu, N.; Sato, Y.; Kainuma, K. Correlation between X-ray diffraction measurements of cellulose crystalline structure and the susceptibility to microbial cellulase. Biotechnol. Bioeng. 1979, 21, 1031–1042. [Google Scholar] [CrossRef]
- Sinitsyn, A.; Gusakov, A.; Vlasenko, E.Y. Effect of structural and physico-chemical features of cellulosic substrates on the efficiency of enzymatic hydrolysis. Appl. Biochem. Biotechnol. 1991, 30, 43–59. [Google Scholar] [CrossRef]
- Nishiyama, Y.; Langan, P.; Chanzy, H. Crystal structure and hydrogen-bonding system in cellulose Iβ from synchrotron X-ray and neutron fiber diffraction. J. Am. Chem. Soc. 2002, 124, 9074–9082. [Google Scholar] [CrossRef]
- Zhu, L.; O’Dwyer, J.P.; Chang, V.S.; Granda, C.B.; Holtzapple, M.T. Structural features affecting biomass enzymatic digestibility. Bioresour. Technol. 2008, 99, 3817–3828. [Google Scholar] [CrossRef] [PubMed]
- Yuan, T.Q.; Wang, W.; Zhang, L.M.; Xu, F.; Sun, R.C. Reconstitution of cellulose and lignin after [C2mim][OAc] pretreatment and its relation to enzymatic hydrolysis. Biotechnol. Bioeng. 2013, 110, 729–736. [Google Scholar] [CrossRef]
- Yang, B.; Dai, Z.; Ding, S.-Y.; Wyman, C.E. Enzymatic hydrolysis of cellulosic biomass. Biofuels 2011, 2, 421–449. [Google Scholar] [CrossRef] [Green Version]
- Huang, Y.-B.; Fu, Y. Hydrolysis of cellulose to glucose by solid acid catalysts. Green Chem. 2013, 15, 1095–1111. [Google Scholar] [CrossRef]
- Hsu, T.-C.; Guo, G.-L.; Chen, W.-H.; Hwang, W.-S. Effect of dilute acid pretreatment of rice straw on structural properties and enzymatic hydrolysis. Bioresour. Technol. 2010, 101, 4907–4913. [Google Scholar] [CrossRef]
- Kafle, K.; Shin, H.; Lee, C.M.; Park, S.; Kim, S.H. Progressive structural changes of Avicel, bleached softwood and bacterial cellulose during enzymatic hydrolysis. Sci. Rep. 2015, 5, 1–10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sacui, I.A.; Nieuwendaal, R.C.; Burnett, D.J.; Stranick, S.J.; Jorfi, M.; Weder, C.; Foster, E.J.; Olsson, R.T.; Gilman, J.W. Comparison of the properties of cellulose nanocrystals and cellulose nanofibrils isolated from bacteria, tunicate, and wood processed using acid, enzymatic, mechanical, and oxidative methods. ACS Appl. Mater. Interfaces 2014, 6, 6127–6138. [Google Scholar] [CrossRef] [PubMed]
- Adams, T.H.; Wieser, J.K.; Yu, J.-H. Asexual sporulation in Aspergillus nidulans. Microbiol. Mol. Biol. Rev. 1998, 62, 35–54. [Google Scholar] [CrossRef] [Green Version]














{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cellulose Sample | CrI (%) | Ihkl Crystallite Size (nm) |
|---|---|---|
| Tunicate cellulose | 93.9 | 9.3 |
| α-cellulose | 70.9 | 2.1 |
| amorphous cellulose | - | - |
| Sample | Hydrolysis Time/h | Mw (g/mol) | DP |
|---|---|---|---|
| tunicate cellulose | 0 | 2.504 × 106 | 4824 |
| 12 | 2.493 × 106 | 4803 | |
| 24 | 2.492 × 106 | 4801 | |
| 48 | 2.489 × 106 | 4795 | |
| 72 | 2.488 × 106 | 4793 | |
| 96 | 2.478 × 106 | 4774 | |
| α-cellulose | 0 | 4.650 × 105 | 896 |
| 12 | 4.489 × 105 | 865 | |
| 24 | 4.437 × 105 | 855 | |
| 48 | 4.255 × 105 | 820 | |
| 72 | 3.778 × 105 | 728 | |
| 96 | 3.684 × 105 | 710 | |
| amorphous cellulose | 0 | 5.906 × 105 | 1138 |
| 1 | 5.475 × 105 | 1055 | |
| 3 | 5.065 × 105 | 976 | |
| 5 | 4.458 × 105 | 859 | |
| 7 | 4.255 × 105 | 820 |
| Before | After | |
|---|---|---|
| Tunicate cellulose | 93.9 | 90.0 |
| α-cellulose | 74.3 | 70.0 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cheng, Y.; Mondal, A.K.; Wu, S.; Xu, D.; Ning, D.; Ni, Y.; Huang, F. Study on the Anti-Biodegradation Property of Tunicate Cellulose. Polymers 2020, 12, 3071. https://doi.org/10.3390/polym12123071
Cheng Y, Mondal AK, Wu S, Xu D, Ning D, Ni Y, Huang F. Study on the Anti-Biodegradation Property of Tunicate Cellulose. Polymers. 2020; 12(12):3071. https://doi.org/10.3390/polym12123071
Chicago/Turabian StyleCheng, Yanan, Ajoy Kanti Mondal, Shuai Wu, Dezhong Xu, Dengwen Ning, Yonghao Ni, and Fang Huang. 2020. "Study on the Anti-Biodegradation Property of Tunicate Cellulose" Polymers 12, no. 12: 3071. https://doi.org/10.3390/polym12123071