Probing Effect of Papirindustriens Forskningsinstitut (PFI) Refining on Aggregation Structure of Cellulose: Crystal Packing and Hydrogen-Bonding Network

Abstract
:1. Introduction
2. Methods
2.1. PFI Refining
2.2. Morphological Analysis of Fibers and Mechanical Properties of Sheets
2.3. Water Retention Value (WRV)
2.4. XRD Analysis
2.5. FT-IR Analysis
3. Results and Discussions
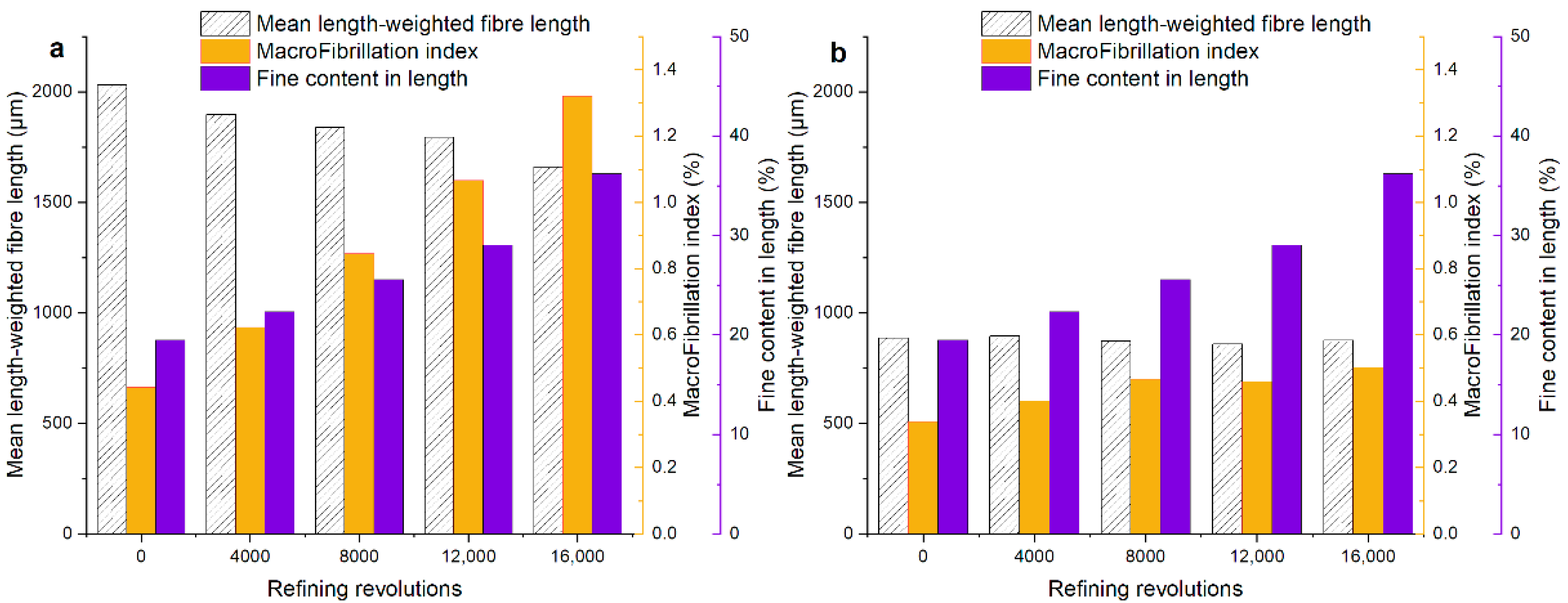
3.1. The Morphological Analysis, Swelling, and Mechanical Properties
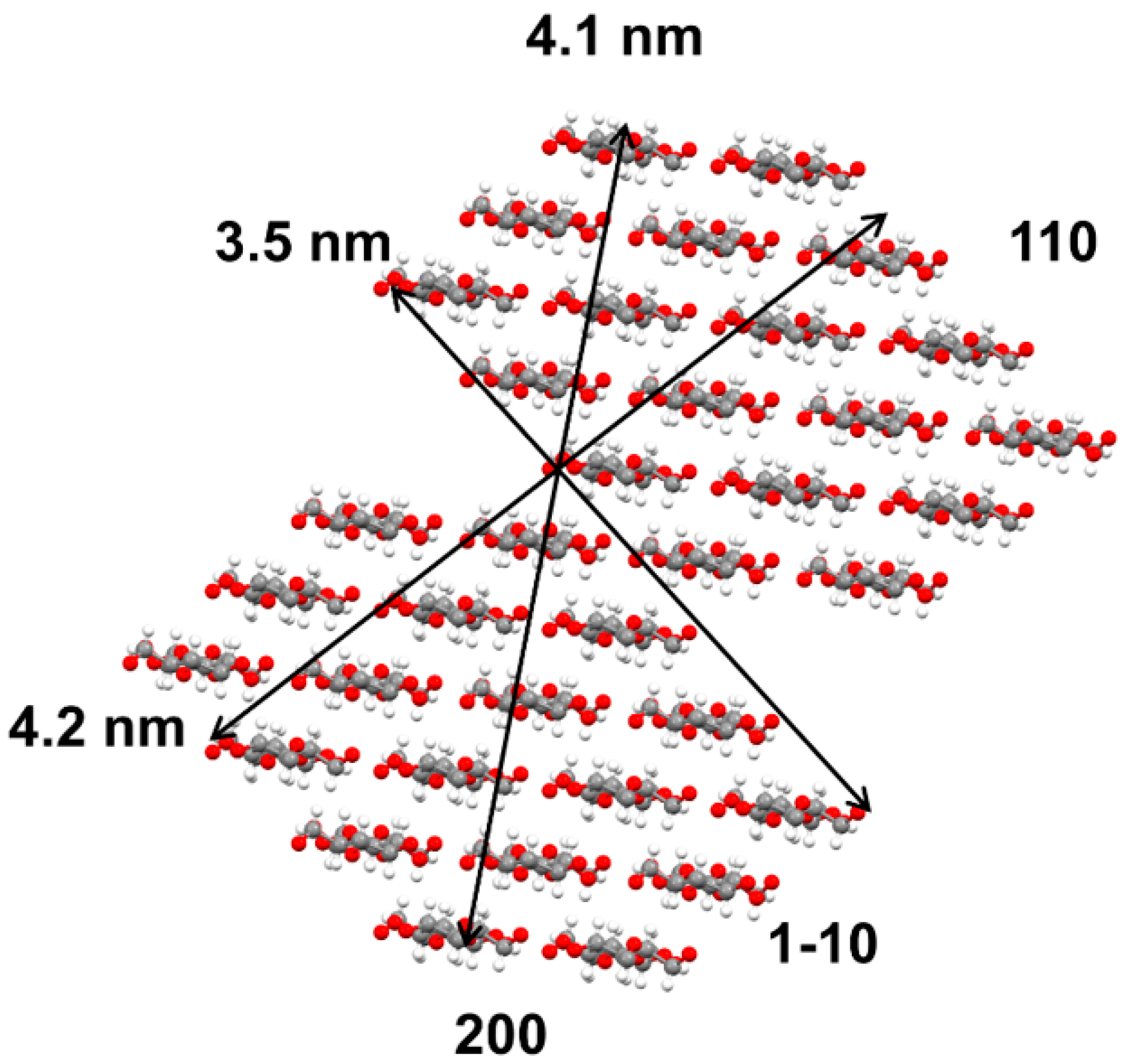
3.2. Effect of PFI Refining on Crystalline Index and Crystallite Size
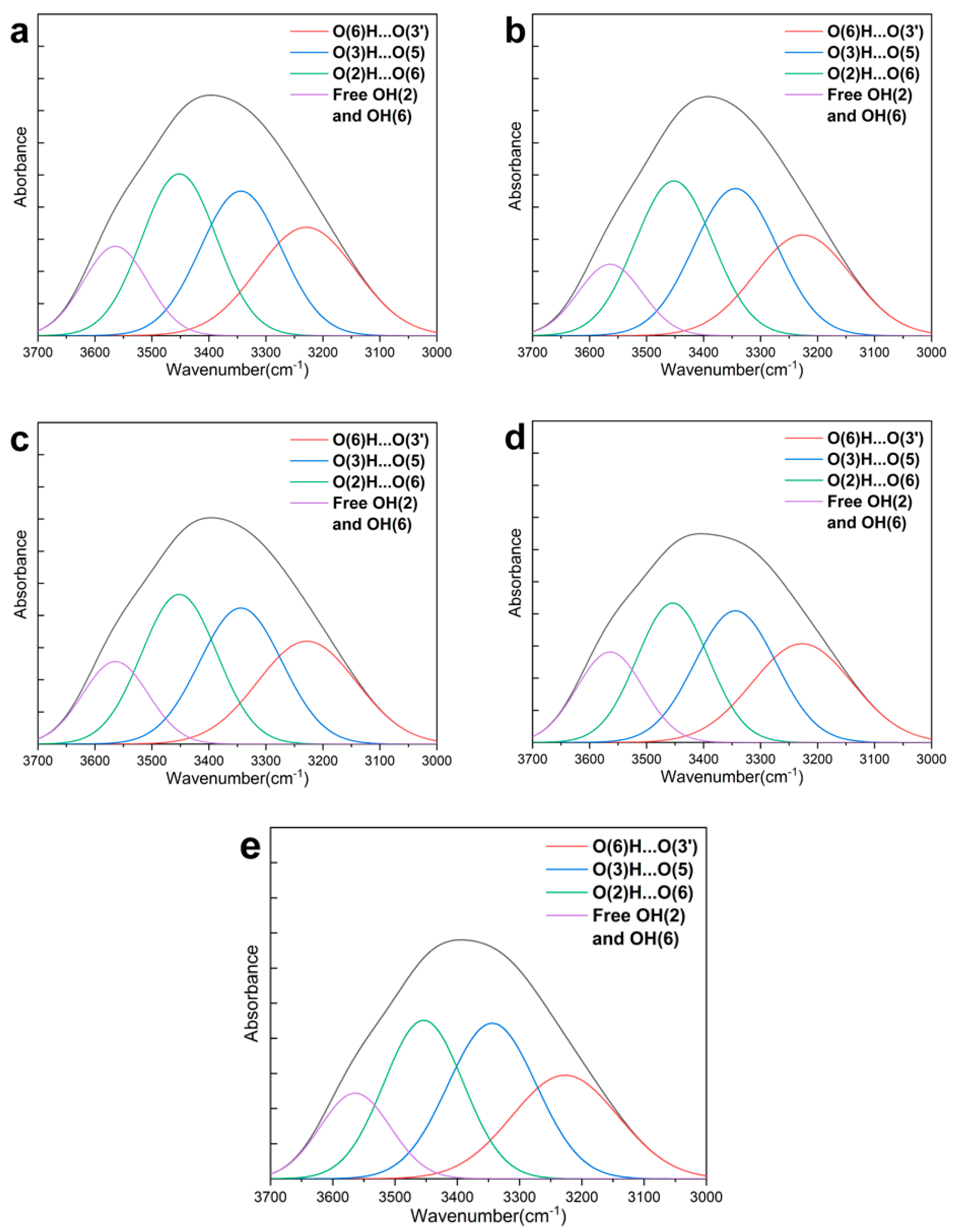
3.3. Effect of PFI Refining on H-Bonding Energies, H-Bonding Distances, and Content of H-Bonds
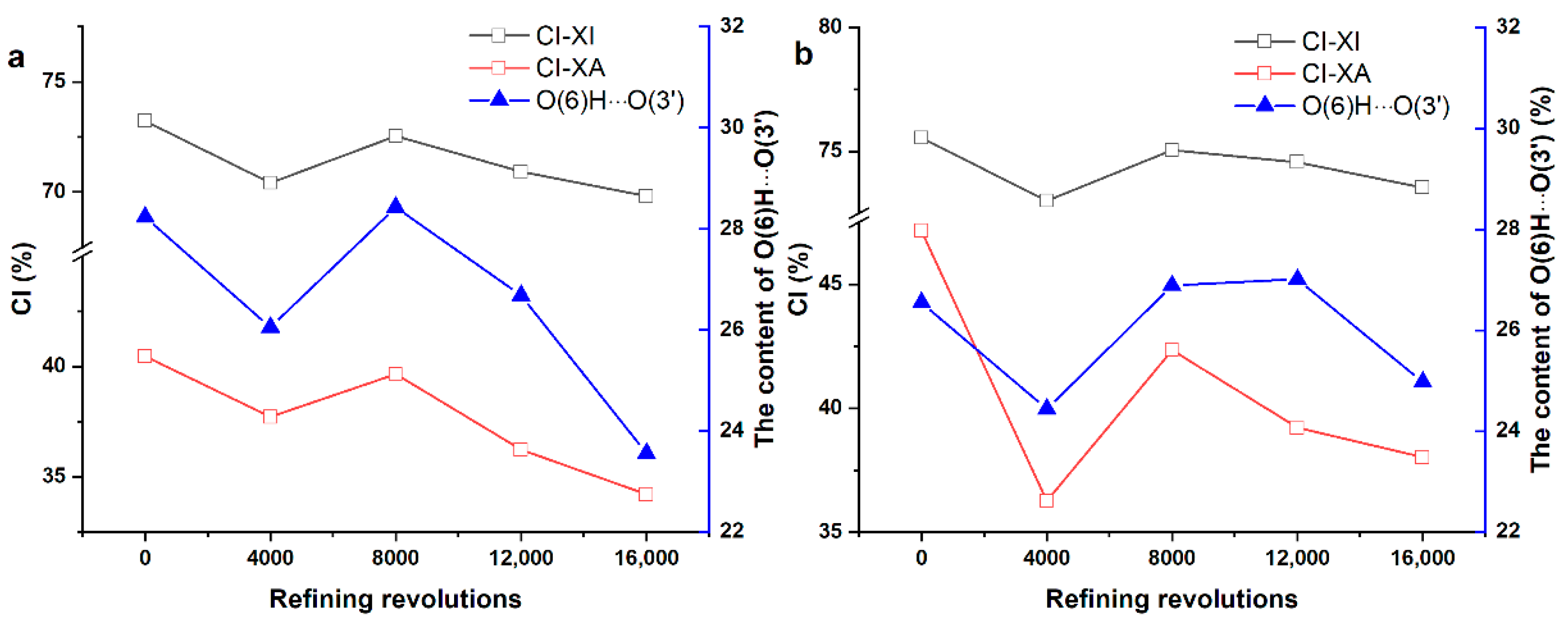
3.4. Correlation between CI and Content of Inter-Molecular H-Bonds during PFI Refining
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Gharehkhani, S.; Sadeghinezhad, E.; Kazi, S.N.; Yarmand, H.; Badarudin, A.; Safaei, M.R.; Zubir, M.N.M. Basic effects of pulp refining on fiber properties—A review. Carbohydr. Polym. 2015, 115, 785–803. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.; Wu, R.; Wang, B.; Hu, Y.; Hou, Q.; Zhang, P.; Wu, R. Comparative study on different pretreatment on enzymatic hydrolysis of corncob residues. Bioresour. Technol. 2020, 295, 122244. [Google Scholar] [CrossRef] [PubMed]
- Lourenço, A.F.; Gamelas, J.A.; Sarmento, P.; Ferreira, P.J. Enzymatic nanocellulose in papermaking–The key role as filler flocculant and strengthening agent. Carbohydr. Polym. 2019, 224, 115200. [Google Scholar] [CrossRef] [PubMed]
- Espinosa, E.; Rol, F.; Bras, J.; Rodríguez, A. Use of multi-factorial analysis to determine the quality of cellulose nanofibers: Effect of nanofibrillation treatment and residual lignin content. Cellulose 2020, 27, 10689–10705. [Google Scholar] [CrossRef]
- Jiménez-López, L.; Eugenio, M.E.; Ibarra, D.; Darder, M.; Martín, J.A.; Martín-Sampedro, R. Cellulose Nanofibers from a Dutch Elm Disease-Resistant Ulmus minor Clone. Polymers 2020, 12, 2450. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Liu, W.; Guan, F.; Li, G.; Song, Z.; Yu, D.; Wang, H.; Liu, H. Using cellulose fibers to fabricate transparent paper by microfibrillation. Carbohydr. Polym. 2019, 214, 26–33. [Google Scholar] [CrossRef] [PubMed]
- Zhao, D.; Yang, F.; Dai, Y.; Tao, F.; Shen, Y.; Duan, W.; Zhou, X.; Ma, H.; Tang, L.; Li, J. Exploring crystalline structural variations of cellulose during pulp beating of tobacco stems. Carbohydr. Polym. 2017, 174, 146–153. [Google Scholar] [CrossRef] [PubMed]
- Zhao, D.; Deng, Y.; Han, D.; Tan, L.; Ding, Y.; Zhou, Z.; Xu, H.; Guo, Y. Exploring structural variations of hydrogen-bonding patterns in cellulose during mechanical pulp refining of tobacco stems. Carbohydr. Polym. 2019, 204, 247–254. [Google Scholar] [CrossRef]
- Xu, H.; Li, B.; Mu, X.; Yu, G.; Liu, C.; Zhang, Y.; Wang, H. Quantitative characterization of the impact of pulp refining on enzymatic saccharification of the alkaline pretreated corn stover. Bioresour. Technol. 2014, 169, 19–26. [Google Scholar] [CrossRef] [PubMed]
- Gao, W.; Xiang, Z.; Chen, K.; Yang, R.; Yang, F. Effect of depth beating on the fiber properties and enzymatic saccharification efficiency of softwood kraft pulp. Carbohydr. Polym. 2015, 127, 400–406. [Google Scholar] [CrossRef]
- Liu, W.; Wang, B.; Hou, Q.; Chen, W.; Wu, M. Effects of fibrillation on the wood fibers’ enzymatic hydrolysis enhanced by mechanical refining. Bioresour. Technol. 2016, 206, 99–103. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Wan, J.; Zhang, X.; Ma, Y.; Wang, Y. Effect of beating on recycled properties of unbleached eucalyptus cellulose fiber. Carbohydr. Polym. 2012, 87, 730–736. [Google Scholar] [CrossRef]
- Popescu, C.; Popescu, M.; Singurel, G.; Vasile, C.; Argyropoulos, D.S.; Willfor, S. Spectral characterization of eucalyptus wood. Appl. Spectrosc. 2007, 61, 1168–1177. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Gao, W.; Chen, K.; Xiang, Z.; Zeng, J.; Wang, B.; Xu, J. Deconstruction of cellulosic fibers to fibrils based on enzymatic pretreatment. Bioresour. Technol. 2018, 267, 426–430. [Google Scholar] [CrossRef] [PubMed]
- Dai, D.; Fan, M. Investigation of the dislocation of natural fibres by Fourier-transform infrared spectroscopy. Vib. Spectrosc. 2011, 55, 300–306. [Google Scholar] [CrossRef]
- Djikanović, D.; Devečerski, A.; Steinbach, G.; Simonović, J.; Matović, B.; Garab, G.; Kalauzi, A.; Radotić, K. Comparison of macromolecular interactions in the cell walls of hardwood, softwood and maize by fluorescence and FTIR spectroscopy, differential polarization laser scanning microscopy and X-ray diffraction. Wood Sci. Technol. 2016, 50, 547–566. [Google Scholar] [CrossRef] [Green Version]
- Popescu, C.M.; Singurel, G.; Popescu, M.C.; Vasile, C.; Argyropoulos, D.S.; Willför, S. Vibrational spectroscopy and X-ray diffraction methods to establish the differences between hardwood and softwood. Carbohydr. Polym. 2009, 77, 851–857. [Google Scholar] [CrossRef]
- Yuan, L.; Wan, J.; Ma, Y.; Wang, Y.; Huang, M.; Chen, Y. The content of different hydrogen bond models and crystal structure of eucalyptus fibers during beating. Bioresources 2012, 8, 717–734. [Google Scholar] [CrossRef] [Green Version]
- Sánchez, P.B.; Tsubaki, S.; Pádua, A.A.; Wada, Y. Kinetic analysis of microwave-enhanced cellulose dissolution in ionic solvents. Phys. Chem. Chem. Phys. 2020, 22, 1003–1010. [Google Scholar] [CrossRef]
- Zhao, L.; Li, S.; Liang, C.; Qiao, L.; Du, K. High-strength and low-crystallinity cellulose/agarose composite microspheres: Fabrication, characterization and protein adsorption. Biochem. Eng. J. 2020, 107826. [Google Scholar] [CrossRef]
- Wang, G.; Yang, X.; Wang, W. Reinforcing linear low-density polyethylene with surfactant-treated microfibrillated cellulose. Polymers 2019, 11, 441. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Elazzouzi-Hafraoui, S.; Nishiyama, Y.; Putaux, J.; Heux, L.; Dubreuil, F.; Rochas, C. The shape and size distribution of crystalline nanoparticles prepared by acid hydrolysis of native cellulose. Biomacromolecules 2008, 9, 57–65. [Google Scholar] [CrossRef] [PubMed]
- Tribulová, T.; Kačík, F.; Evtuguin, D.V.; Čabalová, I.; Ďurkovič, J. The effects of transition metal sulfates on cellulose crystallinity during accelerated ageing of silver fir wood. Cellulose 2019, 26, 2625–2638. [Google Scholar] [CrossRef]
- Kouko, J.; Jajcinovic, M.; Fischer, W.; Ketola, A.; Hirn, U.; Retulainen, E. Effect of mechanically induced micro deformations on extensibility and strength of individual softwood pulp fibers and sheets. Cellulose 2019, 26, 1995–2012. [Google Scholar] [CrossRef] [Green Version]
- Ahvenainen, P.; Kontro, I.; Svedström, K. Comparison of sample crystallinity determination methods by X-ray diffraction for challenging cellulose I materials. Cellulose 2016, 23, 1073–1086. [Google Scholar] [CrossRef] [Green Version]
- Haigler, C.H.; Roberts, A.W. Structure/function relationships in the rosette cellulose synthesis complex illuminated by an evolutionary perspective. Cellulose 2019, 26, 227–247. [Google Scholar] [CrossRef]
- Kubicki, J.D.; Yang, H.; Sawada, D.; O’Neill, H.; Oehme, D.; Cosgrove, D. The shape of native plant cellulose microfibrils. Sci. Rep. 2018, 8, 13983. [Google Scholar] [CrossRef] [Green Version]
- Yang, H.; Kubicki, J.D. A density functional theory study on the shape of the primary cellulose microfibril in plants: Effects of C6 exocyclic group conformation and H-bonding. Cellulose 2020, 27, 2389–2402. [Google Scholar] [CrossRef]
- Duchemin, B.; Thuault, A.; Vicente, A.; Rigaud, B.; Fernandez, C.; Eve, S. Ultrastructure of cellulose crystallites in flax textile fibres. Cellulose 2012, 19, 1837–1854. [Google Scholar] [CrossRef]
- Fernandes, A.N.; Thomas, L.H.; Altaner, C.M.; Callow, P.; Forsyth, V.T.; Apperley, D.C.; Kennedy, C.J.; Jarvis, M.C. Nanostructure of cellulose microfibrils in spruce wood. Proc. Natl. Acad. Sci. USA 2011, 108, E1195–E1203. [Google Scholar] [CrossRef] [Green Version]
- Scholz, D.; Xie, J.; Kröcher, O.; Vogel, F. Mechanochemistry-assisted hydrolysis of softwood over stable sulfonated carbon catalysts in a semi-batch process. RSC Adv. 2019, 9, 33525–33538. [Google Scholar] [CrossRef] [Green Version]
- Li, L.; Zhuang, J.; Zou, H.; Pang, J.; Yu, S. Partition usage of cellulose by coupling approach of supercritical carbon dioxide and cellulase to reducing sugar and nanocellulose. Carbohydr. Polym. 2020, 229, 115533. [Google Scholar] [CrossRef] [PubMed]
- Djahedi, C.; Bergenstråhle-Wohlert, M.; Berglund, L.A.; Wohlert, J. Role of hydrogen bonding in cellulose deformation: The leverage effect analyzed by molecular modeling. Cellulose 2016, 23, 2315–2323. [Google Scholar] [CrossRef]
- Altaner, C.M.; Thomas, L.H.; Fernandes, A.N.; Jarvis, M.C. How cellulose stretches: Synergism between covalent and hydrogen bonding. Biomacromolecules 2014, 15, 791–798. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kondo, T.; Sawatari, C. A Fourier transform infra-red spectroscopic analysis of the character of hydrogen bonds in amorphous cellulose. Polymer 1996, 37, 393–399. [Google Scholar] [CrossRef]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| (a) | |||||
| Revolutions | CI (%) | Crystallite Size (nm) | |||
| CI-XI a | CI-XA b | (110) | (200) | ||
| 0 | 73.23 | 40.46 | 3.2 | 3.5 | 4.5 |
| 4000 | 70.42 | 37.71 | 3.1 | 3.4 | 4.6 |
| 8000 | 72.54 | 39.65 | 3.2 | 3.4 | 4.6 |
| 12,000 | 70.93 | 36.22 | 3.1 | 3.7 | 4.7 |
| 16,000 | 69.82 | 34.20 | 3.2 | 3.5 | 4.7 |
| (b) | |||||
| Revolutions | CI (%) | Crystallite Size (nm) | |||
| CI-XI a | CI-XA b | (110) | (200) | ||
| 0 | 75.55 | 47.16 | 3.1 | 3.1 | 4.5 |
| 4000 | 73.01 | 36.26 | 3.1 | 3.3 | 4.7 |
| 8000 | 75.06 | 42.35 | 3.1 | 3.5 | 4.8 |
| 12,000 | 74.57 | 39.21 | 2.9 | 4.0 | 4.8 |
| 16,000 | 73.55 | 38.01 | 3.0 | 3.7 | 4.7 |
| (a) | |||||
| Revolutions | O(6)H⋯O(3ʹ) | O(3)H⋯O(5) | O(2)H⋯O(6) | Free OH(2) and OH(6) | r2 |
| 0 | 28.23 | 28.44 | 31.40 | 11.93 | 0.9995 |
| 4000 | 26.05 | 31.31 | 32.50 | 10.14 | 0.9995 |
| 8000 | 28.43 | 30.86 | 25.87 | 14.84 | 0.9995 |
| 12,000 | 26.68 | 32.58 | 25.49 | 15.25 | 0.9996 |
| 16,000 | 23.56 | 34.92 | 29.38 | 12.14 | 0.9996 |
| (b) | |||||
| Revolutions | O(6)H⋯O(3ʹ) | O(3)H⋯O(5) | O(2)H⋯O(6) | Free OH(2) and OH(6) | r2 |
| 0 | 26.55 | 29.09 | 30.04 | 14.32 | 0.9990 |
| 4000 | 24.44 | 33.06 | 31.28 | 11.22 | 0.9991 |
| 8000 | 26.89 | 29.81 | 28.99 | 14.31 | 0.9991 |
| 12,000 | 27.01 | 29.28 | 27.40 | 16.31 | 0.9992 |
| 16,000 | 24.98 | 31.65 | 29.85 | 13.52 | 0.9989 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, K.; Zhao, L.; He, B. Probing Effect of Papirindustriens Forskningsinstitut (PFI) Refining on Aggregation Structure of Cellulose: Crystal Packing and Hydrogen-Bonding Network. Polymers 2020, 12, 2912. https://doi.org/10.3390/polym12122912
Li K, Zhao L, He B. Probing Effect of Papirindustriens Forskningsinstitut (PFI) Refining on Aggregation Structure of Cellulose: Crystal Packing and Hydrogen-Bonding Network. Polymers. 2020; 12(12):2912. https://doi.org/10.3390/polym12122912
Chicago/Turabian StyleLi, Kunpeng, Lihong Zhao, and Beihai He. 2020. "Probing Effect of Papirindustriens Forskningsinstitut (PFI) Refining on Aggregation Structure of Cellulose: Crystal Packing and Hydrogen-Bonding Network" Polymers 12, no. 12: 2912. https://doi.org/10.3390/polym12122912