1. Introduction
Polymer crystallization is one of the most important issues in polymer science. Two-thirds of common polymer materials can be crystallized. Controlling the degree of crystallization is the major way to tune the performances of polymer materials, including their mechanical properties, photoelectric properties, thermal conductivity, etc. The mechanism of polymer crystallization are extensively studied. The chain-connectivity makes the problem complex. Although the experimental phenomena have been reported, and the comprehensive theoretical description about polymer crystallization is scarce [
1]. In ideal crystal formed by small molecules, molecules are arranged on an infinite periodic lattice. The polymer chains in their perfect crystals, should be straightened fully, and packed parallelly. This kind of polymer crystal is called as infinite extended chain crystal. Nevertheless, it is impossible for actual polymer chains to organize their configurations in this way to form the perfect crystal in a finite period. With this constrains, the polymer forms folded chain crystal (also called the lamellar crystal) generally [
2,
3,
4]. The stems whose lengths are much shorter than the total contour lengths of polymer chains consist the lamellar crystals. The folded chain segments form the amorphous layers.
In the 1960s, Hoffman and Lauritzen proposed their theory of polymer crystallization, which afterwards was called classical theory [
5,
6,
7,
8,
9]. This theory treats the crystallization behavior as a successive process that the stems attach one by one onto the growth surface. They derived the spontaneous selection of lamellar thickness and the growth rate, and agree with the experimental results in their age [
10]. The prerequisite of this theory is the existing primary nucleus (or a lamellar crystal already exists). In this theory the lamellar has two surface: one is the lateral surface on which the stem deposition happens, and another is the chain-folding surface. In fact, the growth of lamellar crystals both the adhesion of stem on the lateral surface and the increase lamellar thickness by the regulating the conformation of folding chains in amorphous layers. The Lauritzen–Hoffman (L-H) theory pays attention to the successive quasi-equilibrium processes on the lateral surface. Its structure and roles on crystallization have not well addressed.
The folding chain surfaces consist of loops, and a small amount of bridges and tails [
11]. Around this theme there are two contrary mainstream models, adjacent re-entry model proposed by Keller [
2,
12] and revised by Fischer [
3], and switchboard model suggested by Flory [
13,
14]. The former supposes the segments in non-growth surface layer re-enter into the lamellar crystal at the sites which are nearby the location where they project out from the crystal region, while the later supposes the re-entries happen at the sites with arbitrary distances with the projecting-out points. The adjacent re-entry model also divides into two typical schematism. The first one is Keller’s original model [
2] which assumes that the loops re-enter the crystal region after experiencing a short length in interface layer, to form a tight loop. The second one is Fischer’s revised model [
3], which describes the loops with a remarkable longer length, i.e., loose loops. Some other variants or revisions of the models about non-growth surfaces are developed successively, while their key ideas are contained in the above-mentioned models.
Muthukumar used Langevin dynamics and Monte Carlo method to simulate the polymer crystallization to study the primary nucleation, the spontaneous selection of lamellar thickness, and the growth kinetics [
15,
16]. The loop-like conformation formed by chain-folding plays an important role in these aspects. First, the folding surface tension is mainly contributed by the entropy penalty due to formation of the loop conformation. An optimal fraction of chain segments in the crystal can be determined from the balance between free energy gain due to forming the crystal and the entropy penalty due to forming the loop conformation. The entropy penalty can be estimated analytically when two injection points of the loops are assumed to be fixed. This theory had agreed with several experiments. In this theory the distance between two injection points of a loop is assumed to be a constant. This parameter is a length scale that should be optimized. How the injection points are arranged on the surface, and how the loops are packed in the amorphous left are still unclear.
Muthukumar’s theory with an explicit chain model for the amorphous layers, was extended from single-chain system to the multi-chain system by Sommer [
17]. The results indicate that the lamellar thickness increases with the number of chains in the crystal, and extended chain crystals are formed if the number of chains in the crystal is large enough (more than the square of the degree of polymerization of the chains in thermodynamic equilibrium states). Sommer’s multi-chain crystal model relies on two main approximations, i.e., the Gaussian chain statistics and the flat, non-fluctuating folding chain surface. He investigated the loops with finite bending rigidity and the surface with a tilt angle, respectively [
18]. Moreover, it has been pointed that the free energy per loop increases substantially as the end-to-end distance of loops become larger, and reveals the preference of tight loops and close re-entries qualitatively under experimental condition below equilibrium melting temperature. A monotonically increasing relation between free energy per loop and end-to-end distance of loops is obtained by this theory. This is because the bending energy of the chain was ignored. Besides, the quantitative optimal value and the distribution of end-to-end distance are still undetermined.
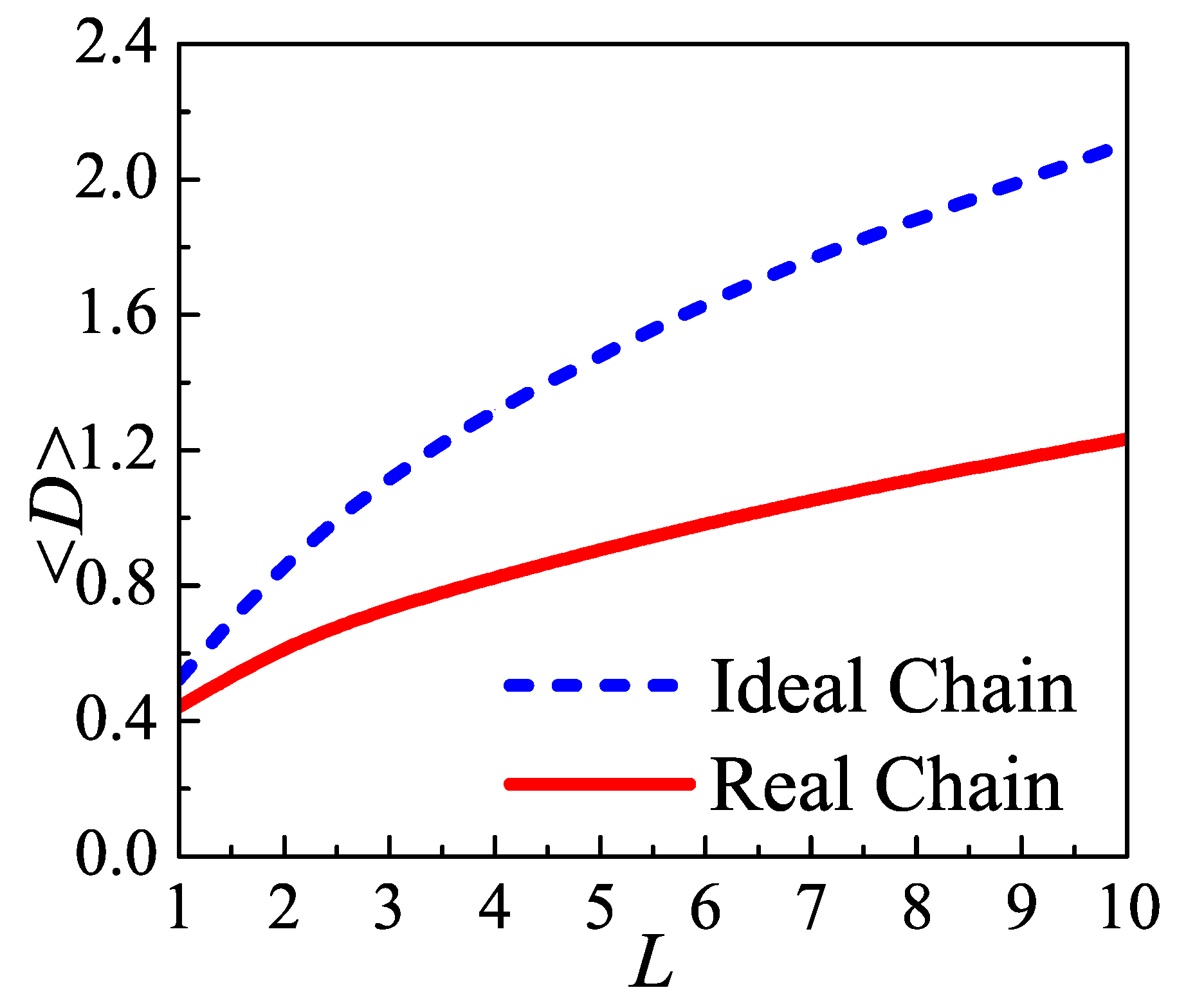
Both Muthukumar’s theory and Sommer’s model indicated that an optimal distance between two injection points exists, which allows the rearrangement of stems and loops after the polymer segments are crystallized. Because two ends of a loop are linked to two stems in lamellar crystal, loops can change their end-to-end distance by the lateral motion of stems, and then minimize the surface tension.
According to the simulation of growth of the lamellar, there are two motion modes: the lateral motion and the parallel motion. The lateral motion (which will be called local-exchange in this paper) is the stem moving in the lateral direction in its entirety. The parallel motion is the reptation motion of the stem, which involves the monomers moving in and out the crystal region. Both motion modes change the loop conformations. The lateral motion changes the end-to-end distance of the loop and the parallel motion change the length of the loops. The simulation work indicated that the lateral motion continues throughout the crystallization process and is even more important than parallel motion during the early stage [
19,
20]. This is just what the L-H theory does not answer. L-H theory is a secondary nucleation theory, in which primary nucleation is already finished [
10].
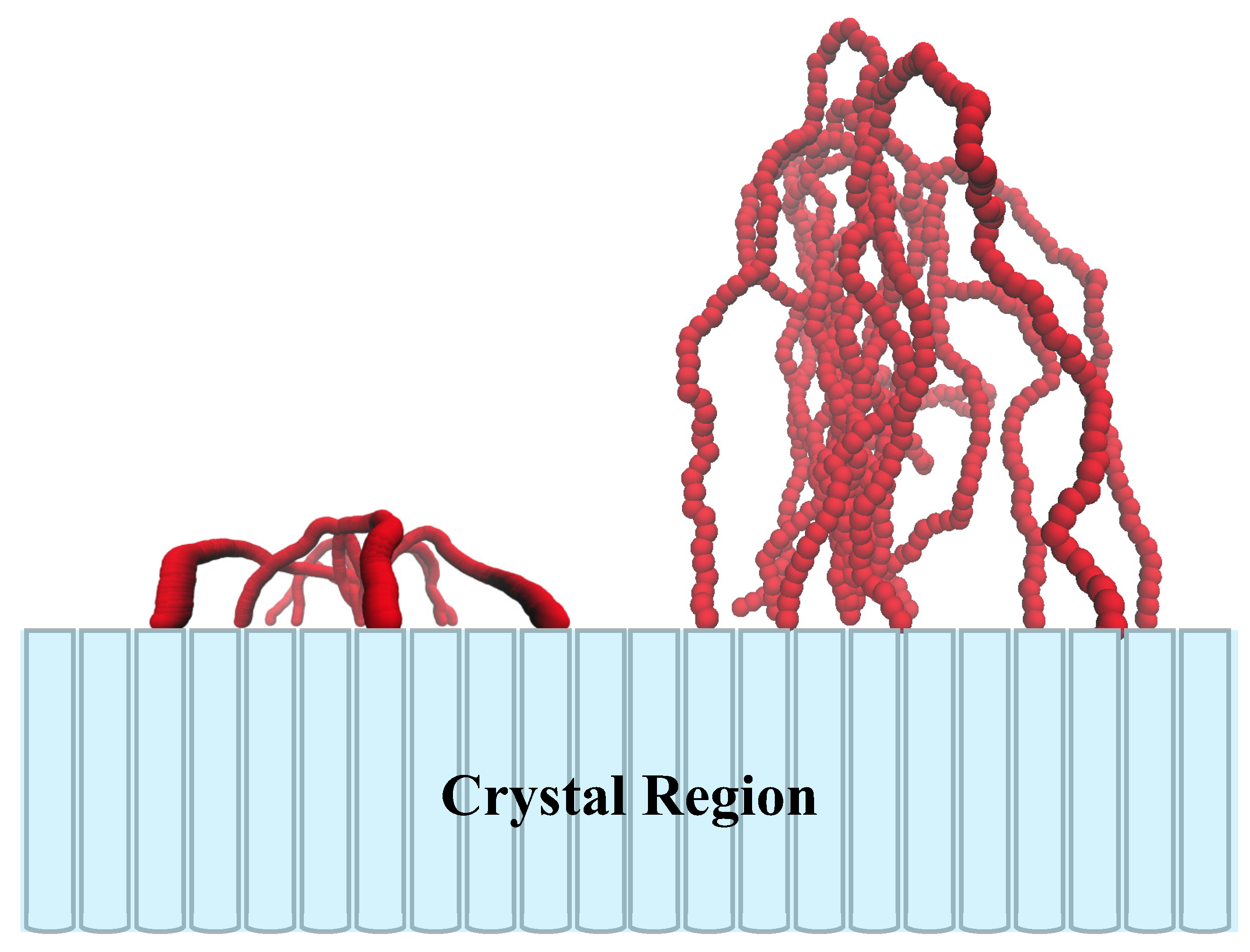
The folding structure of polymer chains in the amorphous layers was observed by Savage et al. recently [
21] using torsional trapping atom force microscopy (AFM). Both many first-neighbor folds and a smaller number of second-neighbor folds are caught. The simulations of interface region can be traced back to Balijepalli’s work in the 1990s [
22,
23,
24,
25]. He modelled the interface layer consisting of loops, tails and bridges by freely rotating chain [
22,
24], and tilt of surface is considered [
25]. Nilsson et al. added two kinds of conformations, free chains and entangled loops, based on Balijepalli’s model, and the hindered rotation model was applied [
26]. The SCFT is introduced to investigate the interface structure by Milner [
27] and Shah [
28]. Because the density of ends is rather low in long polymer systems, Milner modelled the interface by a homogeneous loop-brush in half-space without tail and bridge [
27]. Shah et al considered the crystallization of multi-block copolymers constituted by components with various rigidity. Lempesis et al. simulated the interface between the crystalline and amorphous domains of poly(tetramethylene oxide) by molecular dynamics [
11]. In general, these theoretical models including loops with variable length correspond to parallel motion model. On the other hand, a theoretical model including loops with variable end-to-end distance corresponds to the lateral motion model. Balijepalli gave the density profile of re-entry sites [
22], and Sommer derived the qualitative preference of small end-to-end distance under the equilibrium conditions [
17].
The polymeric loop brush is a helpful theoretical model to characterize the conformation of polymers in the amorphous layer [
27]. The lamellar is considered as the substrate of the polymer brush. The injection points of a loop is modeled as the grafting points of the loop brush. The adjacent re-entry model and the switchboard model of crystallization can be modeled by continuously changing the distance between two injection points.
In the amorphous layer the length of loops are much shorter than the chain length and comparable with that of the Kuhn length. Therefore, its conformation deviates Gaussian statistic behavior and will be dominated by the semiflexible behavior. It has been reported that the end-to-end distance of the semiflexible chain depends on the chain end orientation and the chain length [
29]. Therefore, the loop in the amorphous layer must has an optimal end-to-end distance. The surface tension of the amorphous will depend on the end-to-end distance.
In present work, the lateral motion model of amorphous layer based on the worm-like chain model has been put forward aiming to study the structure of the folding surface. In this model, the two injection points of the loop are allowed to move on the folding surface to model the local-exchange of the stems, which is different from the immobile loop brush [
30], while the influence of the mobility of injection points is neglected. The single-chain in mean-field theory (SCMFT) is used to solve this model, in which the path integral of chain statistics in the auxiliary field is computed by the Monte Carlo simulation [
31,
32,
33]. The conformations of the folded chain in the amorphous region can be well demonstrated by a Wang–Landau algorithm.
This paper is organized as follows: In
Section 2 the model system and the numerical methods are provided which incorporate the Monte Carlo simulation, Wang—Landau algorithm and SCMFT; In
Section 3 the main results of crystal-amorphous interfacial structure have been obtained; In
Section 4 the discussion.
2. Theory and Numerical Method
2.1. Modeling the Crystal-Amorphous Interface
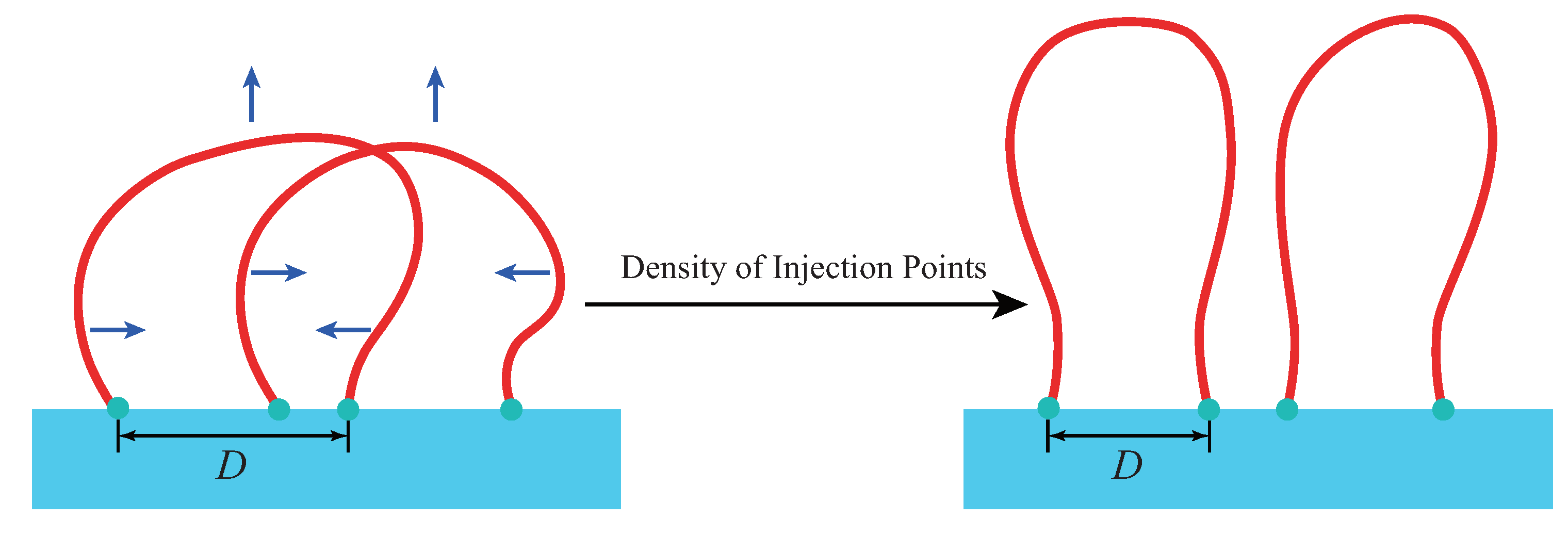
The amorphous layer is considered to be a polymer loop brush with two injection points connecting to two stems in the crystal. The crystal is modeled as the substrate of the brush and the stems are considered implicitly [
10]. Considering stems exchange lateral position in the crystal, the free energy of the crystal region will not be changed. However, the free energy of the amorphous region will be changed due to the variation of the distance between injection points of loops. The parameters of interfacial “pseudo-brush” include (1) the contour length of loop chains, and (2) the areal density of “injected” chains. The latter concerns the lattice constant and is equivalent to “grafting density” in the language of polymer brushes. In above conditions, the system will determines the optimal distance between two injection points via minimizing chain bending and maximizing conformation entropy.
A minimum model is considered here to reveal the lateral motion of the stem to optimize the surface tension of the amorphous layer which is similar to Ref. [
27]. According to the atomistic simulation of poly(tetramethylene oxide), the percentage of loops in the noncrystalline domain is the largest, and much larger than the sum of the corresponding number of bridges and tails [
11], thus, the contribution of tails and bridges can be ignored. To simplify the study, we assume that the direction of stems is normal to the surface of lamellae, although this is not in full accord with the real situations. For example, both experiments [
21,
34] and simulations [
11] point out that there is an angle between the stem and the normal direction of lamellar. Besides, Savage et al. also observed that the lamellae edges have a roughness in the order of nanometers [
21,
24,
25]. The point we focus on here is the minimization of interfacial tension via changing the distance between two injection points. Considering the complexity of this problem, we fixed the loop length, and simplify the system as a mono-disperse loop brush. This simplification ignores the chain-slipping behavior, which will be taken into consideration in our future work.
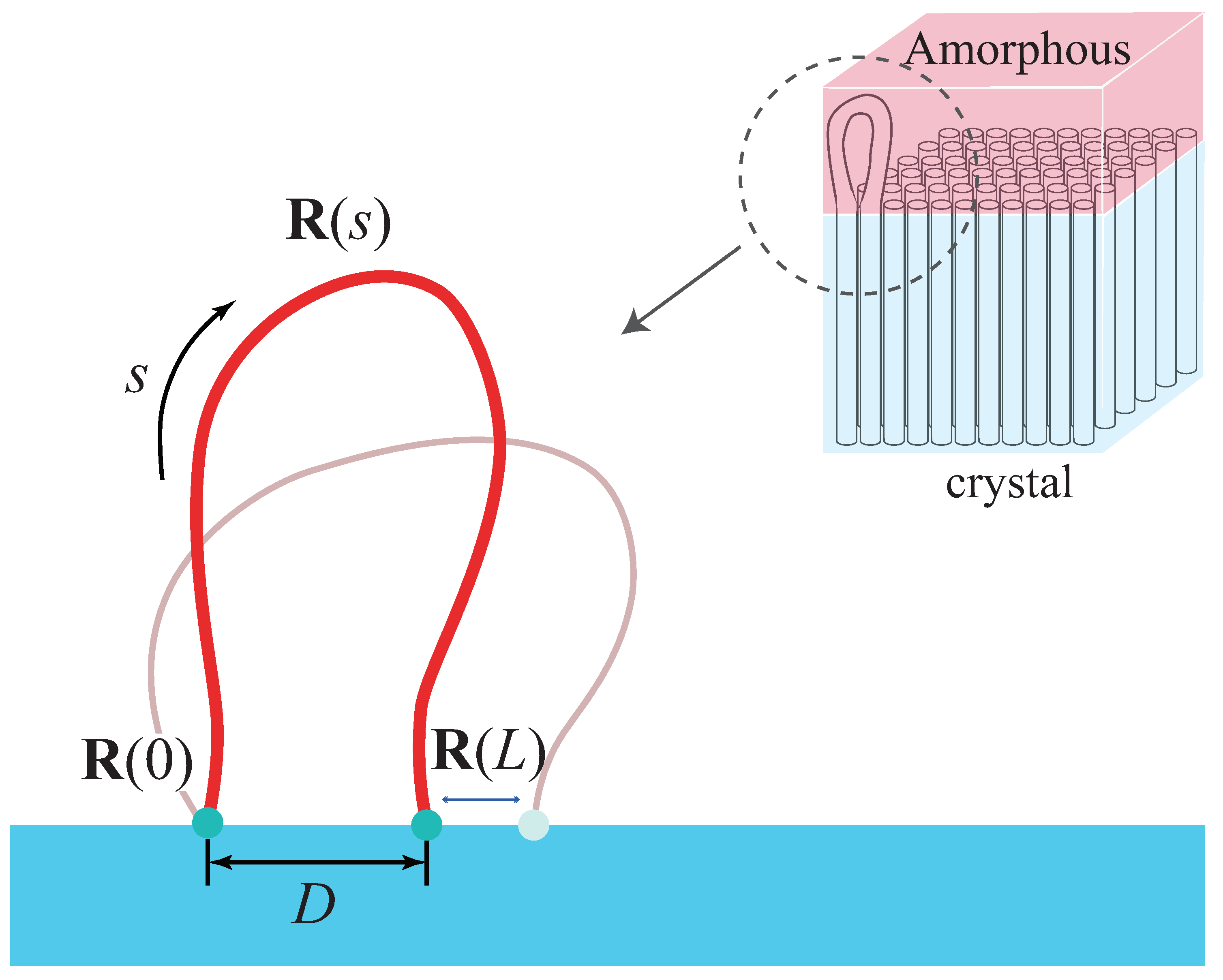
We model the amorphous layer by
n worm-like chains loops with their ends on the lamellar edge (the
plane at
) with area
A, as shown in
Figure 1. The lattice constant is
c, and
is inversely proportional to the density of injection points
[
17,
18]. Each loop has a total contour length
L with persistence length
a and excluded diameter
d. Henceforth, all the lengths are nondimensionalized by
a. The configuration of loop is described by a spacial curve
, where
is the contour variable. The distance between its two injection points is defined by
. The interfacial free energy formula can be given by SCMFT (see
Appendix A for details):
where
is the tangent vector of the chain.
with the Boltzmann factor
and temperature
T.
is the auxiliary field. The
term, which is the single chain partition function, can be written as function of
D (see
Appendix B for details):
where
, defined by Equation (
A15), is the density of states for a given
D;
, defined by Equation (
A21), is the density of energy states for a given
D and
L;
is the ceiling energy. We design a modified Wang–Landau algorithm to accomplish the calculation of
in the ensemble
.
2.2. Modified Multi-Steps High-Precision Wang–Landau Algorithm
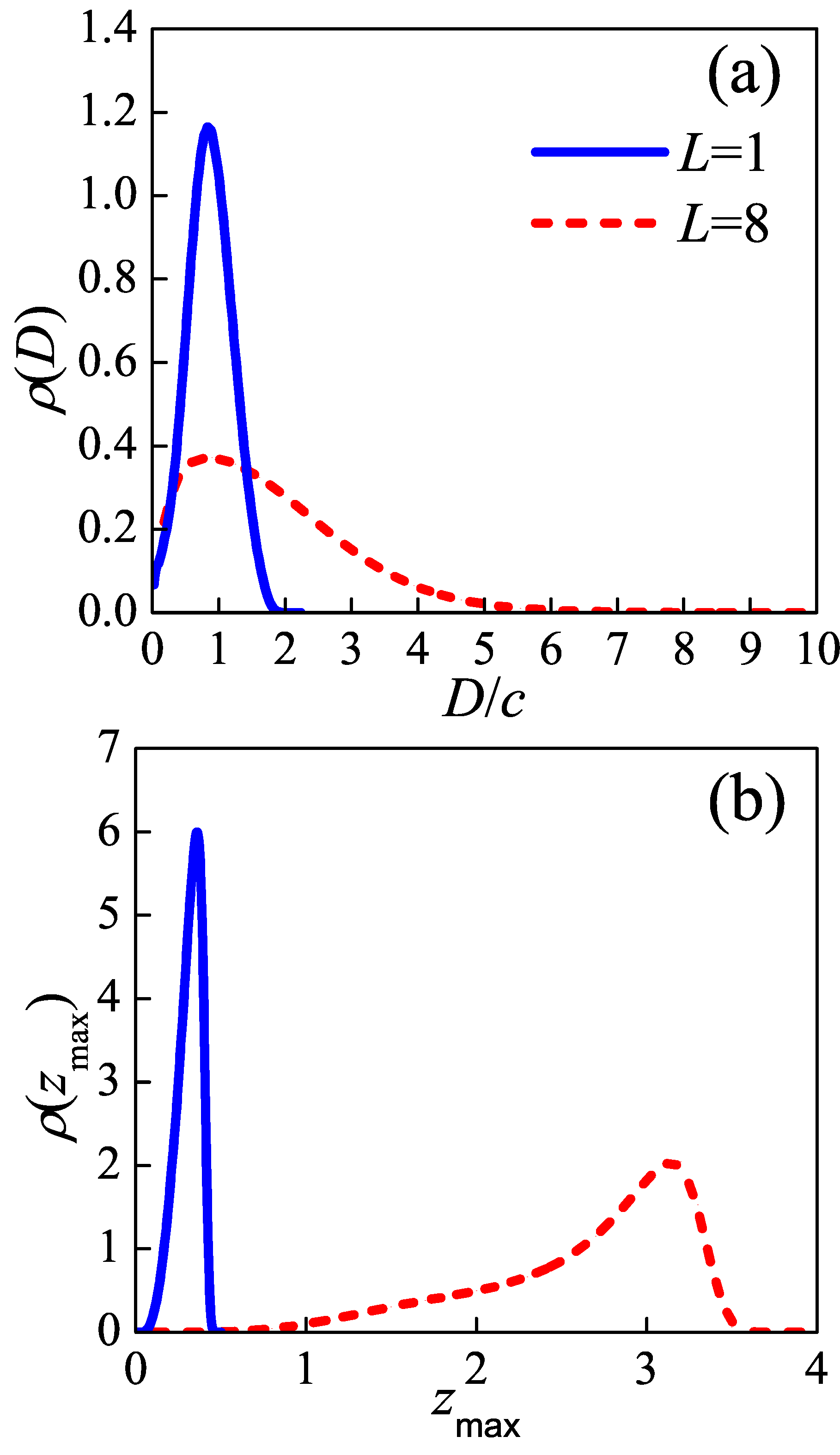
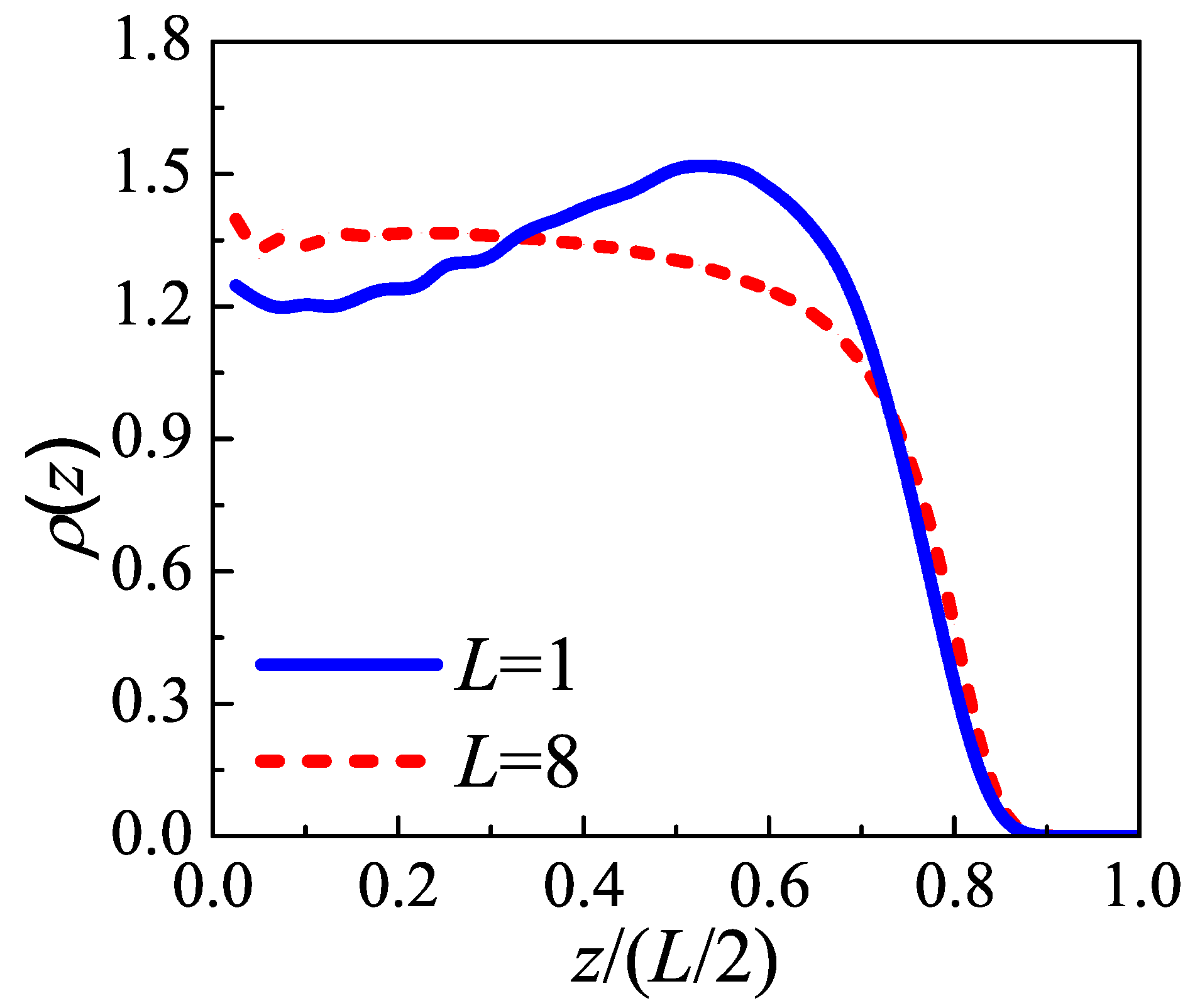
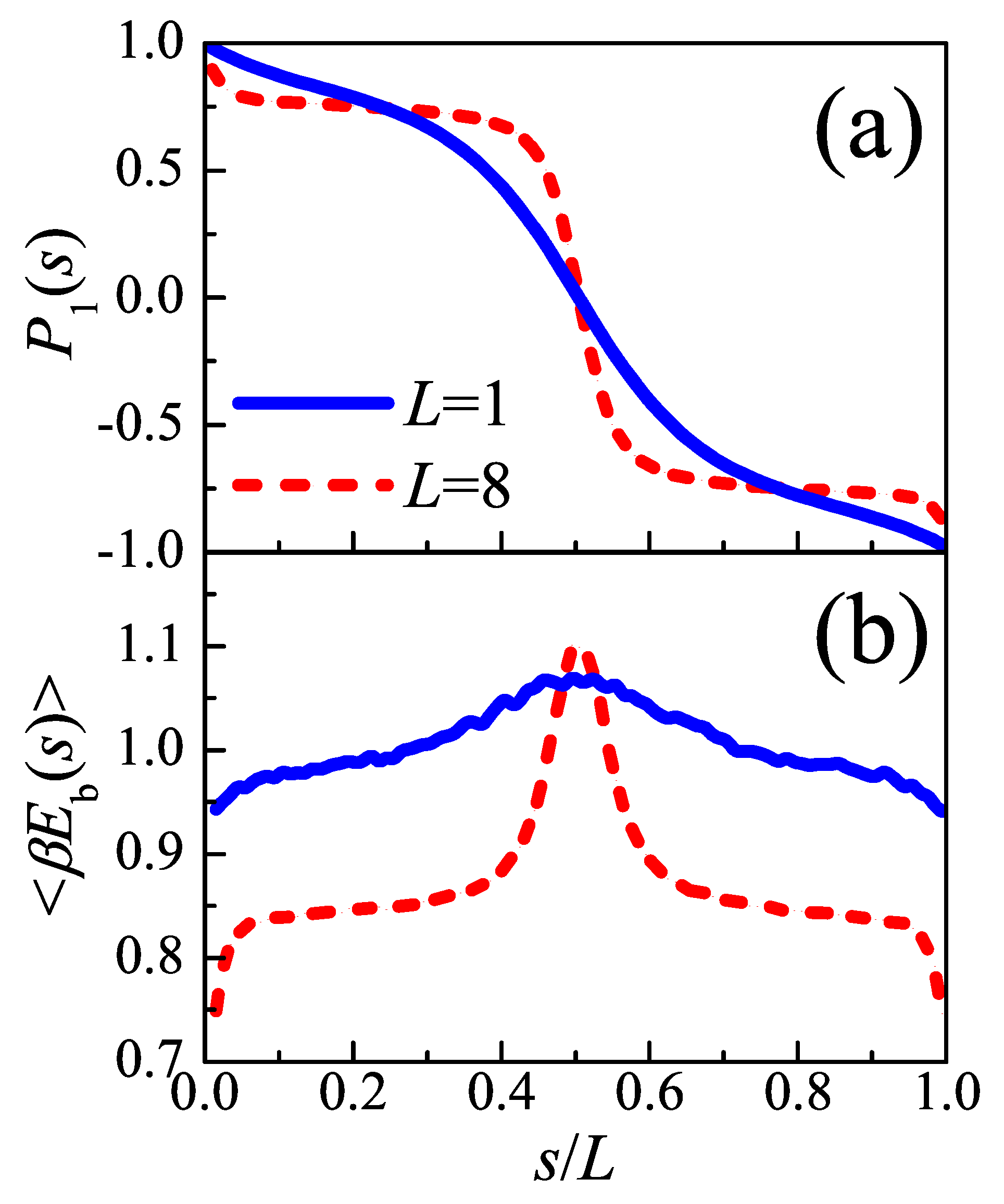
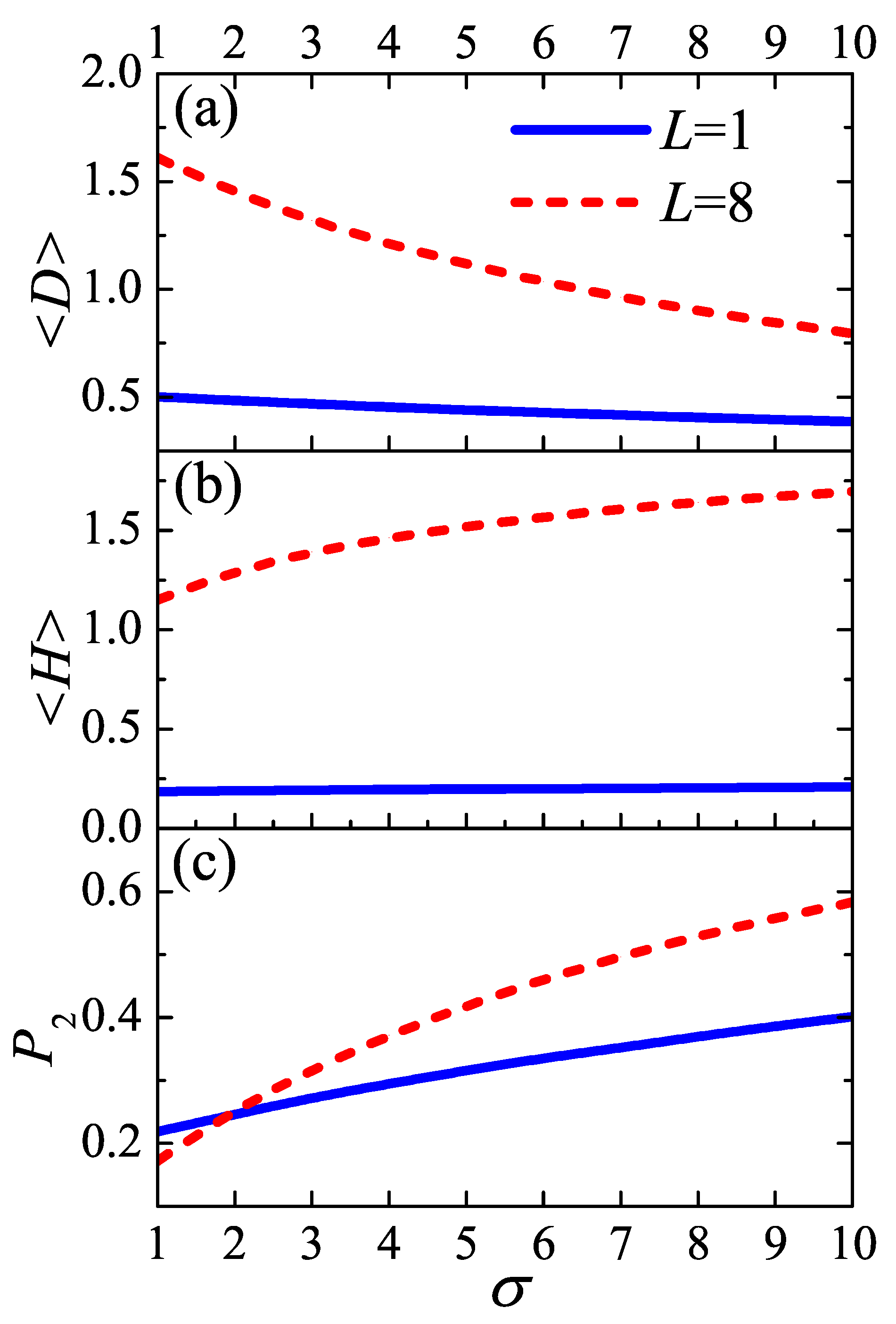
As two typical cases,
and
are chosen to represent the “tight fold” case and the “loose loop” case, respectively. Considering simple cubic polymer crystal, the density of injection points
is inversely proportional to the square of lattice constant
c, i.e.,
. As an example, in the following calculations, we take the persistence length
at the crystallization temperature. With this parameter,
(i.e.,
). For a given auxiliary field
,
Monte Carlo steps are performed to make sure the chain equilibrates under the field and then
conformations are sampled to evaluate the ensemble average of density functional. Then we update the auxiliary field
using the mean-field Equation (
A8) with simple mixing algorithm [
35] to reach the convergent result.
The ensemble average of any observable quantity
is computed by sampling the possible paths in the auxiliary field,
, explicitly by Monte Carlo simulation, i.e.,
where
M is the total number of conformations sampled for the ensemble average and
is the
j-th conformation of a single chain in the auxiliary field sampled by the Metropolis algorithm.
The determination of
written as Equation (
A15) follows the algorithm proposed by Wang and Landau [
36], except that the conventional reference parameter
E is replaced by
D [
37]. The range of variation for
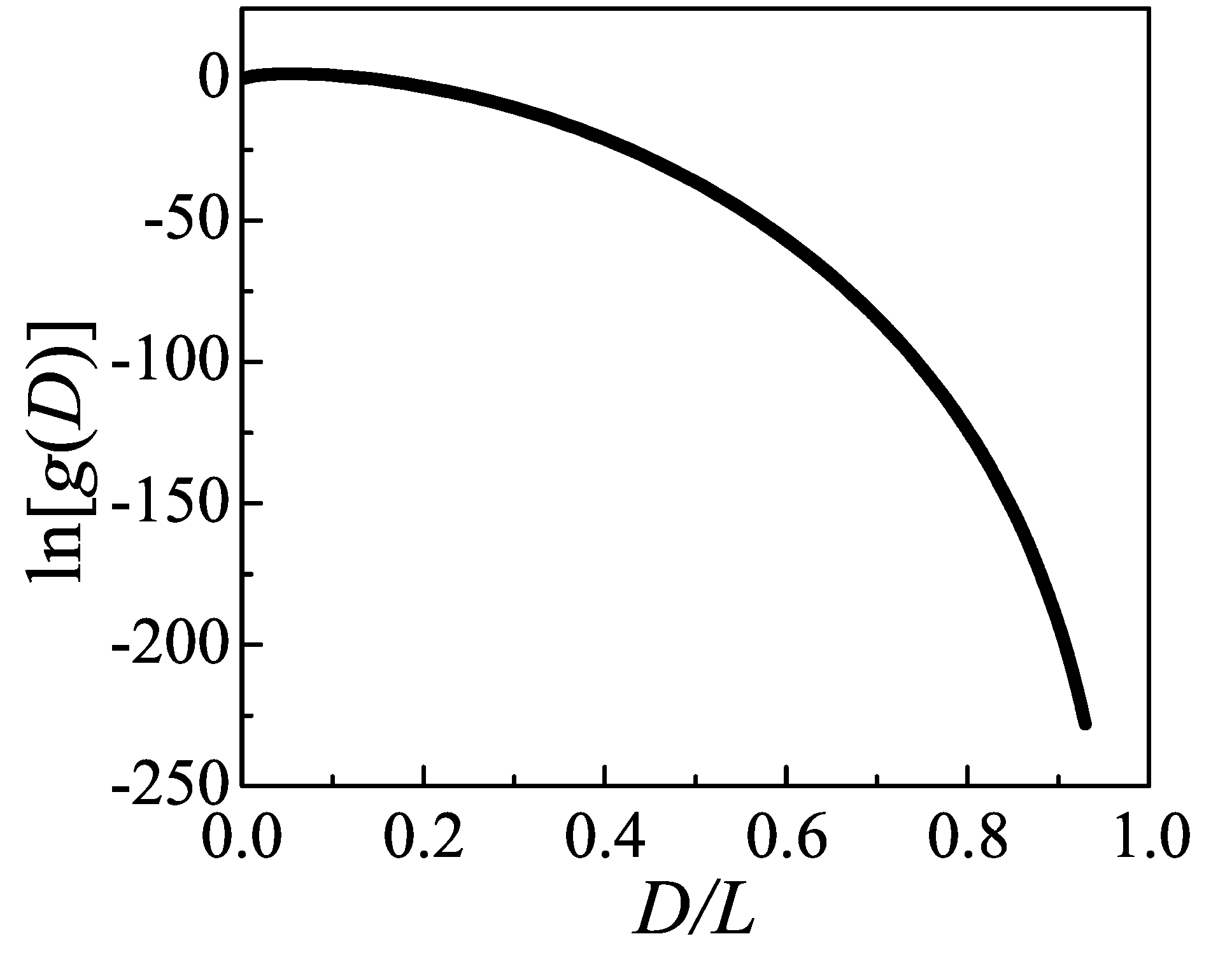
is within
, where
corresponds to the conformation which is fully stretched and clings to the two-dimensional lamellae edge. The
is evenly divided into
equal bins, where the function
for the
i-th bin is represented by a variable
. We take the densities of states
equal to 1 and the accumulating histogram
equal to 0 for all
i in the beginning of procedure, together with a initial modification factor
.
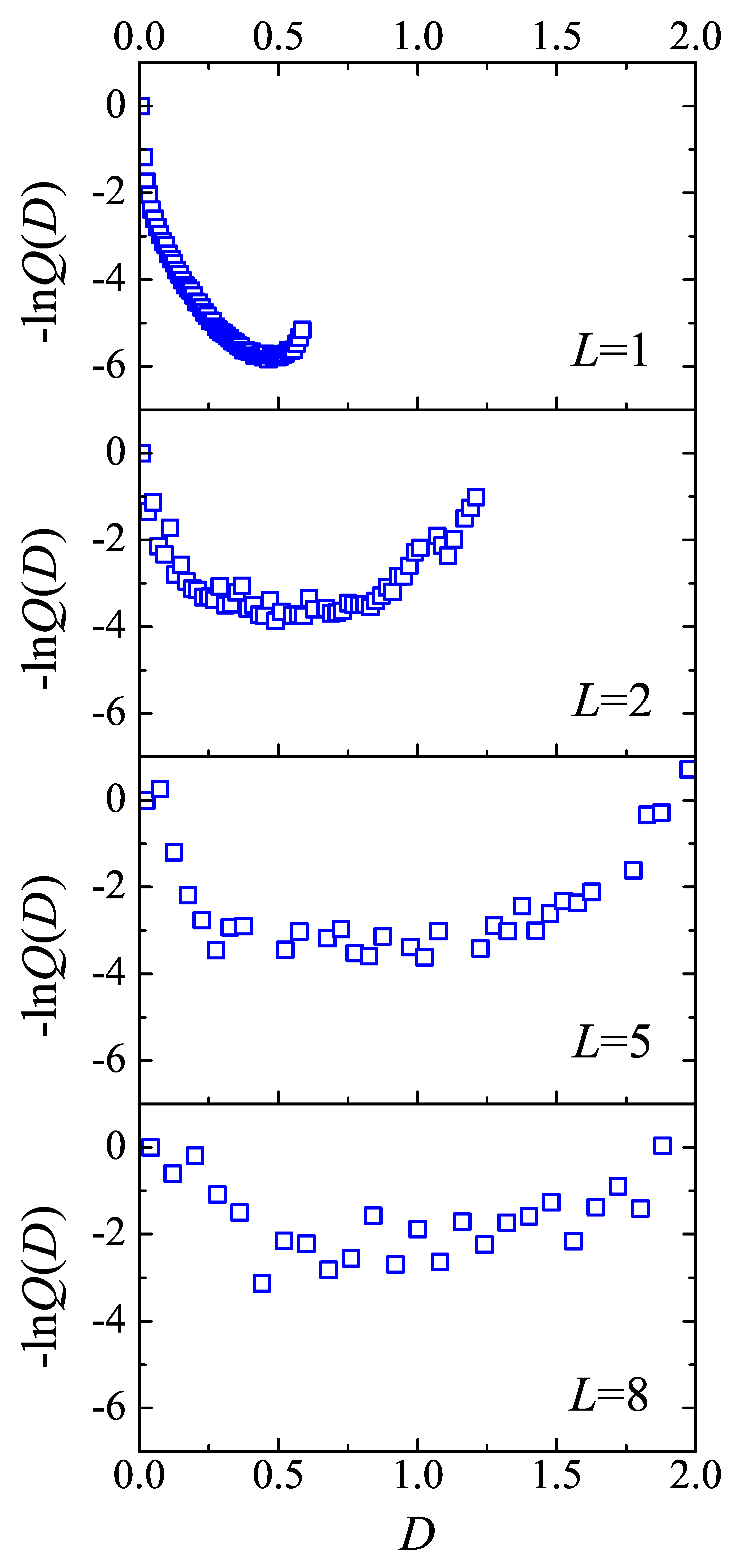
The random walk in D-space is performed by to sample the conformation of inextend loop by Monte Carlo trial move without any consideration of energy. Two trial moves are used in present work: the lateral motion of the injection point and the crankshaft move. In the lateral motion, the segments between a randomly selected segment and a injection point are rotated by a random angle about the axis which is normal the substrate and goes through the selected segment. In the crankshaft move, the segments between randomly selected two segments are rotated by a random angle about the end-to-end vector of these segments. Both trial moves are subject to the hard-wall interaction of the substrate at . The acceptance of the trial move from the conformation with to that with is .
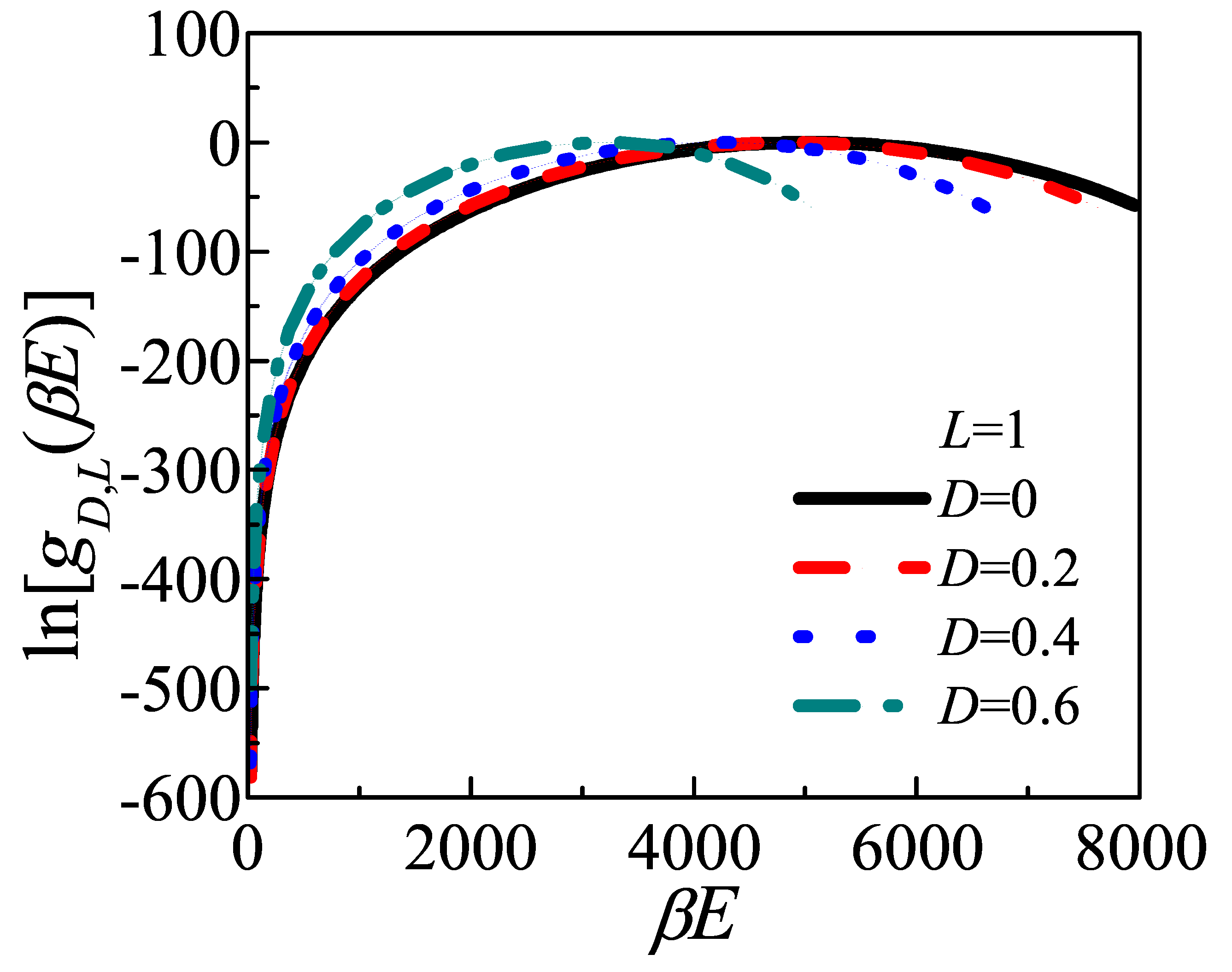
After the Monte Carlo attempt, once the value of D visits the i-th bin, the density of state is updated by the modification factor, i.e., , as well as the accumulating histogram updating by . A simulation iteration terminates when the maximal difference , where is the average of all , is less than . The modification factor, f, is then multiplied by , the histogram is reset to zero, and a new MC simulation iteration was conducted by using the new f until . An approximation for within accuracy no more than has been obtained.
The Wang–Landau algorithm was also used to estimate . In worm-like chain model, the upper bound of bending energy could be as high as for , corresponding to the zigzag conformation. However, the Boltzmann factor would exceed the precision which the computer could achieve for . Considering the integrand is multiplied by and , the precision of density of energy states near ground state is crucial because of the weighting of Boltzmann factor. The conventional Wang–Landau algorithm, could not meet the requirement of precision.
In the view of above-mentioned difficulty, we proposed a modified Wang–Landau scheme to determine the . It contains three steps:
(1) Giving a trial run of Wang–Landau algorithm in the whole energy space, aiming to search a configuration which can be treated as the ground state reasonably.
(2) The E space is divided into regions, and width of every region is of that of the adjacent higher-energy region (in this paper, we take and ). Besides, each region has a small but enough part of overlap with its left and right neighbouring regions, respectively. Wang–Landau algorithm is independently performed in each energy region. A simulation iteration terminates when the maximal difference is less than , and the iterations of MC segments are considered convergent after f become smaller than .
(3) Up to present, the sectional in different energy regions had been obtained independently, but the difference between them is still unknown. To make all of them join together as a smooth curve, we“alining” the overlap of adjacent two regions, which means, shift of all regions till the overlap regions are coincide with their neighbors. This method manages visiting the energy states which are too low for a individual Wang–Landau run to explore.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}










