Comparative Experimental and Theoretical Study of Mg, Al and Zn Aryloxy Complexes in Copolymerization of Cyclic Esters: The Role of the Metal Coordination in Formation of Random Copolymers

 , , , , and
, , , , and
Abstract
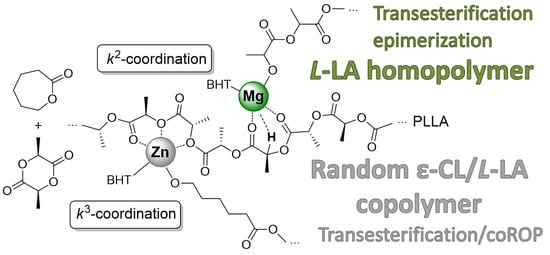
:
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Instruments and Methods
2.3. Preparation and X-ray Diffraction Study of the Complex 4
2.4. Polymerization Experiments
2.4.1. Homopolymerization of lLA and εCL
2.4.2. Copolymerization of lLA and εCL
2.4.3. Synthesis of Random lLA/εCL Copolymers Using PLLA
2.4.4. Copolymerization of εCL with Other Comonomers
2.5. DFT Calculations
3. Results and Discussion
3.1. Homopolymerization of lLA and εCL
3.2. Copolymerization of lLA and εCL
3.3. Synthesis, Molecular Structure and Catalytic Behaviour of the Zn Chelate Complex 4
3.4. Microstructure and Thermal Properties of lLA/εCL Copolymers
3.5. Mechanistic Insights of the Formation of lLA/εCL Copolymers
3.6. Synthesis of Random εCL Copolymers Using PLLA and Other Comonomers
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Jérôme, C.; Lecomte, P. Recent advances in the synthesis of aliphatic polyesters by ring-opening polymerization. Adv. Drug Del. Rev. 2008, 60, 1056–1076. [Google Scholar] [CrossRef]
- Tian, H.; Tang, Z.; Zhuang, X.; Chen, X.; Jing, X. Biodegradable synthetic polymers: Preparation, functionalization and biomedical application. Prog. Polym. Sci. 2012, 37, 237–280. [Google Scholar] [CrossRef]
- Nuyken, O.; Pask, S.D. Ring-Opening Polymerization—An Introductory Review. Polymers 2013, 5, 361–403. [Google Scholar] [CrossRef] [Green Version]
- Lecomte, P.; Jérôme, C. Recent Developments in Ring-Opening Polymerization of Lactones. Adv. Polym. Sci. 2012, 245, 173–218. [Google Scholar] [CrossRef]
- Thomas, C.M. Stereocontrolled ring-opening polymerization of cyclic esters: Synthesis of new polyester microstructures. Chem. Soc. Rev. 2010, 39, 165–173. [Google Scholar] [CrossRef] [PubMed]
- Walsh, D.J.; Hyatt, M.G.; Miller, S.A.; Guironnet, D. Recent Trends in Catalytic Polymerizations. ACS Catal. 2019, 9, 11153–11188. [Google Scholar] [CrossRef]
- Brannigan, R.P.; Dove, A.P. Synthesis, properties and biomedical applications of hydrolytically degradable materials based on aliphatic polyesters and polycarbonates. Biomater. Sci. 2017, 5, 9–21. [Google Scholar] [CrossRef] [PubMed]
- Kamber, N.E.; Jeong, W.; Waymouth, R.M.; Pratt, R.C.; Lohmeijer, B.G.G.; Hedrick, J.L. Organocatalytic Ring-Opening Polymerization. Chem. Rev. 2007, 107, 5813–5840. [Google Scholar] [CrossRef]
- Kiesewetter, M.K.; Shin, E.J.; Hedrick, J.L.; Waymouth, R.M. Organocatalysis: Opportunities and Challenges for Polymer Synthesis. Macromolecules 2010, 43, 2093–2107. [Google Scholar] [CrossRef]
- Fevre, M.; Pinaud, J.; Gnanou, Y.; Vignolle, J.; Taton, D. N-Heterocyclic carbenes (NHCs) as organocatalysts and structural components in metal-free polymer synthesis. Chem. Soc. Rev. 2013, 42, 2142–2172. [Google Scholar] [CrossRef]
- Dove, A.P. Organic Catalysis for Ring-Opening Polymerization. ACS Macro Lett. 2012, 1, 1409–1412. [Google Scholar] [CrossRef]
- Hu, S.; Zhao, J.; Zhang, G.; Schlaad, H. Macromolecular architectures through organocatalysis. Prog. Polym. Sci. 2017, 74, 34–77. [Google Scholar] [CrossRef]
- Khalil, A.; Cammas-Marion, S.; Coulembier, O. Organocatalysis Applied to the Ring-Opening Polymerization of β-Lactones: A Brief Overview. J. Polym. Sci. Part A Polym. Chem. 2019, 57, 657–672. [Google Scholar] [CrossRef]
- Bossion, A.; Heifferon, K.V.; Meabe, L.; Zivic, N.; Taton, D.; Hedrick, J.L.; Long, T.E.; Sardon, H. Opportunities for organocatalysis in polymer synthesis via step-growth methods. Prog. Polym. Sci. 2019, 90, 164–210. [Google Scholar] [CrossRef]
- Nifant’ev, I.; Ivchenko, P. DFT Modeling of Organocatalytic Ring-Opening Polymerization of Cyclic Esters: A Crucial Role of Proton Exchange and Hydrogen Bonding. Polymers 2019, 11, 2078. [Google Scholar] [CrossRef] [Green Version]
- Sarazin, Y.; Carpentier, J.-F. Discrete Cationic Complexes for Ring-Opening Polymerization Catalysis of Cyclic Esters and Epoxides. Chem. Rev. 2015, 115, 3564–3614. [Google Scholar] [CrossRef]
- Jianming, R.; Anguo, X.; Hongwei, W.; Hailin, Y. Review—Recent development of ring-opening polymerization of cyclic esters using aluminum complexes. Design. Monomers Polym. 2014, 17, 345–355. [Google Scholar] [CrossRef] [Green Version]
- Kremer, A.B.; Mehrkhodavandi, P. Dinuclear catalysts for the ring opening polymerization of lactide. Coord. Chem. Rev. 2019, 380, 35–57. [Google Scholar] [CrossRef]
- Lyubov, D.M.; Tolpygin, A.O.; Trifonov, A.A. Rare-earth metal complexes as catalysts for ring-opening polymerization of cyclic esters. Coord. Chem. Rev. 2019, 392, 83–145. [Google Scholar] [CrossRef]
- Nifant’ev, I.; Ivchenko, P. Coordination Ring-Opening Polymerization of Cyclic Esters: A Critical Overview of DFT Modeling and Visualization of the Reaction Mechanisms. Molecules 2019, 24, 4117. [Google Scholar] [CrossRef] [Green Version]
- Zhang, X.; Fevre, M.; Jones, G.O.; Waymouth, R.M. Catalysis as an Enabling Science for Sustainable Polymers. Chem. Rev. 2018, 118, 839–885. [Google Scholar] [CrossRef] [PubMed]
- Arbaoui, A.; Redshaw, C. Metal catalysts for ε-caprolactone polymerisation. Polym. Chem. 2010, 1, 801–826. [Google Scholar] [CrossRef]
- Zhong, Z.; Dijkstra, P.J.; Feijen, J. Controlled synthesis of biodegradable lactide polymers and copolymers using novel in situ generated or single-site stereoselective polymerization initiators. J. Biomater. Sci. Polym. Ed. 2004, 15, 929–946. [Google Scholar] [CrossRef] [PubMed]
- Hillmyer, M.A.; Tolman, W.B. Aliphatic polyester block polymers: Renewable, degradable, sustainable. Acc. Chem. Res. 2014, 47, 2390–2396. [Google Scholar] [CrossRef]
- Albertsson, A.-C.; Varma, I.K. Recent Developments in Ring Opening Polymerization of Lactones for Biomedical Applications. Biomacromolecules 2003, 4, 1466–1486. [Google Scholar] [CrossRef]
- Slomkowski, S. Biodegradable Polyesters for Tissue Engineering. Macromol. Symp. 2007, 253, 47–58. [Google Scholar] [CrossRef]
- Naira, L.S.; Laurencin, C.T. Biodegradable polymers as biomaterials. Prog. Polym. Sci. 2007, 32, 762–798. [Google Scholar] [CrossRef]
- Vert, M. Aliphatic Polyesters: Great Degradable Polymers That Cannot Do Everything. Biomacromolecules 2005, 6, 538–546. [Google Scholar] [CrossRef]
- Maitz, M.F. Applications of synthetic polymers in clinical medicine. Biosurface Biotribology 2015, 1, 161–176. [Google Scholar] [CrossRef] [Green Version]
- Pappalardo, D.; Mathisen, T.; Finne-Wistrand, A. Biocompatibility of resorbable polymers: A historical perspective and framework for the future. Biomacromolecules 2019, 20, 1465–1477. [Google Scholar] [CrossRef] [Green Version]
- Woodruff, M.A.; Hutmacher, D.W. The return of a forgotten polymer—Polycaprolactone in the 21st century. Prog. Polym. Sci. 2010, 35, 1217–1256. [Google Scholar] [CrossRef] [Green Version]
- Sisson, A.L.; Ekinci, D.; Lendlein, A. The contemporary role of ε-caprolactone chemistry to create advanced polymer architectures. Polymer 2013, 54, 4333–4350. [Google Scholar] [CrossRef] [Green Version]
- Liu, W.; Feng, Z.; Ou-Yang, W.; Pan, X.; Wang, X.; Huang, P.; Zhang, C.; Kong, D.; Wang, W. 3D printing of implantable elastic PLCL copolymer scaffolds. Soft Matter 2020, 16, 2141–2148. [Google Scholar] [CrossRef] [PubMed]
- Ruengdechawiwat, S.; Somsunan, R.; Molloy, R.; Siripitayananon, J.; Franklin, V.J.; Topham, P.D.; Tighe, B.J. Controlled Synthesis and Processing of a Poly(L-lactide-co-ε-caprolactone) Copolymer for Biomedical Use as an Absorbable Monofilament Surgical Suture. Adv. Mater. Res. 2014, 894, 172–176. [Google Scholar] [CrossRef]
- Fernández, J.; Etxeberria, A.; Sarasua, J.-R. Synthesis, structure and properties of poly(L-lactide-co-ε-caprolactone) statistical copolymers. J. Mechan. Behavior Biomed. Mater. 2012, 9, 100–112. [Google Scholar] [CrossRef]
- Fernández, J.; Larrañaga, A.; Etxeberria, A.; Wang, W.; Sarasua, J.R. A new generation of poly(lactide/ε-caprolactone) polymeric biomaterials for application in the medical field. J. Biomed. Mater. Res. Part A 2014, 102, 3573–3584. [Google Scholar] [CrossRef]
- Ruengdechawiwat, S.; Siripitayananon, J.; Molloy, R.; Somsunan, R.; Topham, P.D.; Tighe, B.J. Preparation of a poly(L-lactide-co-caprolactone) copolymer using a novel tin(II) alkoxide initiator and its fiber processing for potential use as an absorbable monofilament surgical suture. Int. J. Polym. Mat. Polym. Biomat. 2016, 65, 277–284. [Google Scholar] [CrossRef] [Green Version]
- Phetsuk, S.; Molloy, R.; Nalampang, K.; Meepowpan, P.; Topham, P.D.; Tighe, B.J.; Punyodom, W. Physical and thermal properties of l-lactide/ϵ-caprolactone copolymers: The role of microstructural design. Polym. Int. 2020, 69, 248–256. [Google Scholar] [CrossRef]
- Vion, J.M.; Jerome, R.; Teyssie, P.; Aubin, M.; Prudhomme, R.E. Synthesis, characterization, and miscibility of caprolactone random copolymers. Macromolecules 1986, 19, 1828–1838. [Google Scholar] [CrossRef]
- Vanhoorne, P.; Dubois, P.; Jerome, R.; Teyssie, P. Macromolecular engineering of polylactones and polylactides. 7. Structural analysis of copolyesters of ε-caprolactone and L- or D,L-lactide initiated by triisopropoxyaluminum. Macromolecules 1992, 25, 37–44. [Google Scholar] [CrossRef]
- Choi, E.-J.; Park, J.-K.; Chang, H.-N. Effect of polymerization catalysts on the microstructure of P(L-LA-co -εCL). J. Polym. Sci. Patr B Polym. Phys. 1994, 32, 2481–2489. [Google Scholar] [CrossRef]
- Matsubara, K.; Eda, K.; Ikutake, Y.; Dan, M.; Tanizaki, N.; Koga, Y.; Yasuniwa, M. Aluminum complex initiated copolymerization of lactones and DL-lactide to form crystalline gradient block copolymers containing stereoblock lactyl chains. J. Polym. Sci. Part A Polym. Chem. 2016, 54, 2536–2544. [Google Scholar] [CrossRef]
- Bai, J.; Xiao, X.; Zhang, Y.; Chao, J.; Chen, X. β-Pyridylenolate zinc catalysts for the ring-opening homo- and copolymerization of ε-caprolactone and lactides. Dalton Trans. 2017, 46, 9846–9858. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, H.-C.; Li, Z.-J.; Wang, B.; Chen, X.; Li, Y.-S. Synthesis of lactide/ɛ-caprolactone quasi-random copolymer by using rationally designed mononuclear aluminum complexes with modified β-ketiminato ligand. J. Polym. Sci. Part A Polym. Chem. 2018, 56, 203–212. [Google Scholar] [CrossRef]
- Harinath, A.; Bhattacharjee, J.; Sarkar, A.; Nayek, H.P.; Panda, T.K. Ring Opening Polymerization and Copolymerization of Cyclic Esters Catalyzed by Group 2 Metal Complexes Supported by Functionalized P–N Ligands. Inorg. Chem. 2018, 57, 2503–2516. [Google Scholar] [CrossRef]
- Wang, Y.; Xiang, D.; Ren, R.; Luo, J.; Sun, W.; Shen, Z. L-lactide homopolymerization and copolymerization with ɛ-caprolactone by tetrahydrosalen stabilized yttrium borohydride complex. Chin. J. Polym. Sci. 2014, 32, 1500–1506. [Google Scholar] [CrossRef]
- Nifant’ev, I.; Shlyakhtin, A.; Kosarev, M.; Gavrilov, D.; Karchevsky, S.; Ivchenko, P. DFT Visualization and Experimental Evidence of BHT-Mg-Catalyzed Copolymerization of Lactides, Lactones and Ethylene Phosphates. Polymers 2019, 11, 1641. [Google Scholar] [CrossRef] [Green Version]
- Florczak, M.; Duda, A. Effect of the Configuration of the Active Center on Comonomer Reactivities: The Case of ε-Caprolactone/l,l-Lactide Copolymerization. Angew. Chem. Int. Ed. 2008, 47, 9088–9091. [Google Scholar] [CrossRef]
- Li, G.; Lamberti, M.; Pappalardo, D.; Pellecchia, C. Random Copolymerization of ε-Caprolactone and Lactides Promoted by Pyrrolylpyridylamido Aluminum Complexes. Macromolecules 2012, 45, 8614–8620. [Google Scholar] [CrossRef]
- Nomura, N.; Akita, A.; Ishii, R.; Mizuno, M. Random Copolymerization of ε-Caprolactone with Lactide Using a Homosalen−Al Complex. J. Am. Chem. Soc. 2010, 132, 1750–1751. [Google Scholar] [CrossRef]
- Wang, Y.; Ma, H. Exploitation of dinuclear salan aluminum complexes for versatile copolymerization of ε-caprolactone and L-lactide. Chem. Commun. 2012, 6729–6731. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Liu, B.; Liu, D.; Wu, C.; Li, S.; Liu, B.; Cui, D. Copolymerization of ε-Caprolactone and l-Lactide Catalyzed by Multinuclear Aluminum Complexes: An Immortal Approach. Organometallics 2014, 33, 6474–6480. [Google Scholar] [CrossRef]
- Sun, Z.; Duan, R.; Yang, J.; Zhang, H.; Li, S.; Pang, X.; Chen, W.; Chen, X. Bimetallic Schiff base complexes for stereoselective polymerisation of racemic-lactide and copolymerisation of racemic-lactide with ε-caprolactone. RSC Adv. 2016, 6, 17531–17538. [Google Scholar] [CrossRef]
- Kan, C.; Ma, H. Copolymerization of L-lactide and ε-caprolactone catalyzed by mono-and dinuclear salen aluminum complexes bearing bulky 6,6′-dimethylbipheyl-bridge: Random and tapered copolymer. RSC Adv. 2016, 6, 47402–47409. [Google Scholar] [CrossRef]
- Pilone, A.; De Maio, N.; Press, K.; Venditto, V.; Pappalardo, D.; Mazzeo, M.; Pellecchia, C.; Kol, M.; Lamberti, M. Ring-opening homo- and co-polymerization of lactides and ε-caprolactone by salalen aluminum complexes. Dalton Trans. 2015, 44, 2157–2165. [Google Scholar] [CrossRef]
- Yang, J.; Sun, Z.; Duan, R.; Li, L.; Pang, X.; Chen, X. Copolymer of lactide and ε-caprolactone catalyzed by bimetallic Schiff base aluminum complexes. Sci. China Chem. 2016, 59, 1384–1389. [Google Scholar] [CrossRef]
- Chandanabodhi, D.; Nanok, T. A DFT study of the ring-opening polymerization mechanism of l-lactide and ε-caprolactone using aluminium salen-type initiators: Towards an understanding of their reactivities in homo- and copolymerization. Mol. Catal. 2017, 436, 145–156. [Google Scholar] [CrossRef]
- Meléndez, D.O.; Castro-Osma, J.A.; Lara-Sánchez, A.; Rojas, R.S.; Otero, A. Ring-opening polymerization and copolymerization of cyclic esters catalyzed by amidinate aluminum complexes. J. Polym. Sci. Part A Polym. Chem. 2017, 55, 2397–2407. [Google Scholar] [CrossRef]
- Shi, T.; Luo, W.; Liu, S.; Li, Z. Controlled random copolymerization of rac-lactide and ɛ-caprolactone by well-designed phenoxyimine Al complexes. J. Polym. Sci. Part A Polym. Chem. 2018, 56, 611–617. [Google Scholar] [CrossRef]
- García-Valle, F.M.; Cuenca, T.; Mosquera, M.E.G.; Milione, S.; Cano, J. Ring-Opening Polymerization (ROP) of cyclic esters by a versatile aluminum Diphenoxyimine Complex: From polylactide to random copolymers. Eur. Polym. J. 2020, 125, 109527. [Google Scholar] [CrossRef]
- Chumsaeng, P.; Haesuwannakij, S.; Virachotikul, A.; Phomphrai, K. Random copolymerization of l-lactide and ε-caprolactone by aluminum alkoxide complexes supported by N2O2 bis(phenolate)-amine ligands. J. Polym. Sci. Part A Polym. Chem. 2019, 57, 1635–1644. [Google Scholar] [CrossRef]
- Honrado, M.; Otero, A.; Fernández-Baeza, J.; Sánchez-Barba, L.F.; Garcés, A.; Lara-Sánchez, A.; Rodríguez, A.M. Copolymerization of Cyclic Esters Controlled by Chiral NNO-Scorpionate Zinc Initiators. Organometallics 2016, 35, 189–197. [Google Scholar] [CrossRef]
- Lin, L.; Xu, Y.; Wang, S.; Xiao, M.; Meng, Y. Ring-opening polymerization of l-lactide and ε-caprolactone catalyzed by versatile tri-zinc complex: Synthesis of biodegradable polyester with gradient sequence structure. Eur. Polym. J. 2016, 74, 109–119. [Google Scholar] [CrossRef]
- Wyrębiak, R.; Oledzka, E.; Figat, R.; Sobczak, M. Application of Diethylzinc/Propyl Gallate Catalytic System for Ring-Opening Copolymerization of rac-Lactide and ε-Caprolactone. Molecules 2019, 24, 4168. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fang, C.; Ma, H. Ring-opening polymerization of rac-lactide, copolymerization of rac-lactide and ε-caprolactone by zinc complexes bearing pyridyl-based tridentate amino-phenolate ligands. Eur. Polym. J. 2019, 119, 289–297. [Google Scholar] [CrossRef]
- Keram, M.; Ma, H. Ring-opening polymerization of lactide, ε-caprolactone and their copolymerization catalyzed by β-diketiminate zinc complexes. Appl. Organomet. Chem. 2017, 31, e3893. [Google Scholar] [CrossRef]
- Webster, R.L.; Noroozi, N.; Hatzikiriakos, S.G.; Thomson, J.A.; Schafer, L.L. Titanium pyridonates and amidates: Novel catalysts for the synthesis of random copolymers. Chem. Commun. 2013, 49, 57–59. [Google Scholar] [CrossRef]
- Lapenta, R.; Mazzeo, M.; Grisi, F. Monoamidinate titanium complexes: Highly active catalysts for the polymerization and copolymerization of L-lactide and ε-caprolactone. RSC Adv. 2015, 5, 87635–87644. [Google Scholar] [CrossRef]
- Gilmour, D.J.; Webster, R.L.; Perrya, M.R.; Schafer, L.L. Titanium pyridonates for the homo- and copolymerization of rac-lactide and ε-caprolactone. Dalton Trans. 2015, 44, 12411–12419. [Google Scholar] [CrossRef] [Green Version]
- Pappuru, S.; Chakraborty, D.; Sundar, J.V.; Roymuhury, S.K.; Ramkumar, V.; Subramanian, V.; Chand, D.K. Group 4 complexes of salicylbenzoxazole ligands as effective catalysts for the ring-opening polymerization of lactides, epoxides and copolymerization of ε-caprolactone with L-lactide. Polymer 2016, 102, 231–247. [Google Scholar] [CrossRef]
- Della Monica, F.; Luciano, E.; Buonerba, A.; Grassi, A.; Milione, S.; Capacchione, C. Poly(lactide-co-ε-caprolactone) copolymers prepared using bis-thioetherphenolate group 4 metal complexes: Synthesis, characterization and morphology. RSC Adv. 2014, 4, 51262–51267. [Google Scholar] [CrossRef]
- Dakshinamoorthy, D.; Peruch, F. Block and random copolymerization of ε-caprolactone, L-, and rac-lactide using titanium complex derived from aminodiol ligand. J. Polym. Sci. Part A Polym. Chem. 2012, 50, 2161–2171. [Google Scholar] [CrossRef]
- Sun, Z.; Zhao, Y.; Santoro, O.; Elsegood, M.R.J.; Bedwell, E.V.; Zahra, K.; Walton, A.; Redshaw, C. Use of titanocalix[4]arenes in the ring opening polymerization of cyclic esters. Catal. Sci. Technol. 2020, 10, 1619–1639. [Google Scholar] [CrossRef]
- Maruta, Y.; Abiko, A. Random copolymerization of ε-caprolactone and l-lactide with molybdenum complexes. Polym. Bull. 2014, 71, 989–999. [Google Scholar] [CrossRef]
- Slattery, R.M.; Stahl, A.E.; Brereton, K.R.; Rheingold, A.L.; Green, D.B.; Fritsch, J.M. Ring opening polymerization and copolymerization of L-lactide and ɛ-caprolactone by bis-ligated magnesium complexes. J. Polym. Sci. Part A Polym. Chem. 2019, 57, 48–59. [Google Scholar] [CrossRef] [Green Version]
- Shao, J.; Zhou, H.; Wang, Y.; Luo, Y.; Yao, Y. Lanthanum complexes stabilized by a pentadentate Schiff-base ligand: Synthesis, characterization, and reactivity in statistical copolymerization of ε-caprolactone and l-lactide. Dalton Trans. 2020, 49, 5842–5850. [Google Scholar] [CrossRef]
- Fadlallah, S.; Jothieswaran, J.; Capet, F.; Bonnet, F.; Visseaux, M. Mixed Allyl Rare-Earth Borohydride Complexes: Synthesis, Structure, and Application in (Co-)Polymerization Catalysis of Cyclic Esters. Chem. Eur. J. 2017, 23, 15644–15654. [Google Scholar] [CrossRef]
- Ouyang, H.; Yuan, D.; Nie, K.; Zhang, Y.; Yao, Y.; Cui, D. Synthesis and Characterization of Dinuclear Salan Rare-Earth Metal Complexes and Their Application in the Homo- and Copolymerization of Cyclic Esters. Inorg. Chem. 2018, 57, 9028–9038. [Google Scholar] [CrossRef]
- Chen, Y.-H.; Chen, Y.-J.; Tseng, H.-C.; Lian, C.-J.; Tsai, H.-Y.; Lai, Y.-C.; Hsu, S.C.N.; Chiang, M.Y.; Chen, H.-Y. Comparing L-lactide and ε-caprolactone polymerization by using aluminum complexes bearing ketiminate ligands: Steric, electronic, and chelating effects. RSC Adv. 2015, 5, 100272–100280. [Google Scholar] [CrossRef]
- Gutmann, V. Solvent effects on the reactivities of organometallic compounds. Coord. Chem. Rev. 1976, 18, 225–255. [Google Scholar] [CrossRef]
- Nifant’ev, I.E.; Shlyakhtin, A.V.; Bagrov, V.V.; Minyaev, M.E.; Churakov, A.V.; Karchevsky, S.G.; Birin, K.P.; Ivchenko, P.V. Mono-BHT heteroleptic magnesium complexes: Synthesis, molecular structure and catalytic behavior in the ring-opening polymerization of cyclic esters. Dalton Trans. 2017, 46, 12132–12146. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Balasanthiran, V.; Chisholm, M.H.; Choojun, K.; Durr, C.B. Ethyl 2-hydroxy-2-methylpropanoate derivatives of magnesium and zinc. The effect of chelation on the homo- and copolymerization of lactide and ε-caprolactone. Dalton Trans. 2014, 43, 2781–2788. [Google Scholar] [CrossRef] [PubMed]
- Chumsaeng, P.; Haesuwannakij, S.; Bureekaew, S.; Ervithayasuporn, V.; Namuangruk, S.; Phomphrai, K. Polymerization of ε-Caprolactone Using Bis(phenoxy)-amine Aluminum Complex: Deactivation by Lactide. Inorg. Chem. 2018, 57, 10170–10179. [Google Scholar] [CrossRef] [PubMed]
- Nifant’ev, I.E.; Shlyakhtin, A.V.; Tavtorkin, A.N.; Kosarev, M.A.; Gavrilov, D.E.; Komarov, P.D.; Ilyin, S.O.; Karchevsky, S.G.; Ivchenko, P.V. Mechanistic study of transesterification in TBD-catalyzed ring-opening polymerization of methyl ethylene phosphate. Eur. Polym. J. 2019, 118, 393–403. [Google Scholar] [CrossRef]
- Kricheldorf, H.R.; Bornhorst, K.; Hachmann-Thiessen, H. Bismuth(III) n-Hexanoate and Tin(II) 2-Ethylhexanoate Initiated Copolymerizations of ε-Caprolactone and l-Lactide. Macromolecules 2005, 38, 5017–5024. [Google Scholar] [CrossRef]
- Fernández, J.; Meaurio, E.; Chaos, A.; Etxeberria, A.; Alonso-Varona, A.; Sarasua, J.R. Synthesis and characterization of poly (l-lactide/ε-caprolactone) statistical copolymers with well resolved chain microstructures. Polymer 2013, 54, 2621–2631. [Google Scholar] [CrossRef]
- Bonné, C.; Pahwa, A.; Picard, C.; Visseaux, M. Bismuth tris-silylamide: A new non-toxic metal catalyst for the ring opening (co-)polymerization of cyclic esters under smooth conditions. Inorg. Chim. Acta 2017, 455, 521–527. [Google Scholar] [CrossRef]
- Balasanthiran, V.; Beilke, T.L.; Chisholm, M.H. Use of over the counter oral relief aids or dietary supplements for the ring-opening polymerization of lactide. Dalton Trans. 2013, 42, 9274–9278. [Google Scholar] [CrossRef]
- Ouyang, H.; Nie, K.; Yuan, D.; Zhang, Y.; Cui, D.; Yao, Y. A convenient method to prepare random LA/CL copolymers from poly(L-lactide) and ε-caprolactone. Sci. China Chem. 2018, 61, 708–714. [Google Scholar] [CrossRef]
- González, V.; Vignati, D.A.L.; Pons, M.-N.; Montarges-Pelletier, E.; Bojic, C.; Giamberini, L. Lanthanide ecotoxicity: First attempt to measure environmental risk for aquatic organisms. Environ. Pollut. 2015, 199, 139–147. [Google Scholar] [CrossRef]
- Herrmann, H.; Nolde, J.; Berger, S.; Heise, S. Aquatic ecotoxicity of lanthanum—A review and an attempt to derive water and sediment quality criteria. Ecotoxic. Environ. Safety 2016, 124, 213–238. [Google Scholar] [CrossRef] [Green Version]
- Cui, J.; Zhang, Z.; Bai, W.; Zhang, L.; He, X.; Ma, Y.; Liu, Y.; Chai, Z. Effects of rare earth elements La and Yb on the morphological and functional development of zebrafish embryos. J. Environ. Sci. 2012, 24, 209–213. [Google Scholar] [CrossRef]
- Chen, H.-Y.; Mialon, L.; Abboud, K.A.; Miller, S.A. Comparative Study of Lactide Polymerization with Lithium, Sodium, Magnesium, and Calcium Complexes of BHT. Organometallics 2012, 31, 5252–5261. [Google Scholar] [CrossRef]
- Fang, H.-J.; Lai, P.-S.; Chen, J.-Y.; Hsu, S.C.N.; Peng, W.-D.; Ou, S.-W.; Lai, Y.-C.; Chen, Y.-J.; Chung, H.; Chen, Y.; et al. ε-Caprolactone polymerization under air by the biocatalyst: Magnesium 2,6-di-tert-butyl-4-methylphenoxide. J. Polym. Sci. Part A Polym. Chem. 2012, 50, 2697–2704. [Google Scholar] [CrossRef]
- Wilson, J.A.; Hopkins, S.A.; Wright, P.M.; Dove, A.P. ‘Immortal’ ring-opening polymerization of ω-pentadecalactone by Mg(BHT)2(THF)2. Polym. Chem. 2014, 5, 2691–2694. [Google Scholar] [CrossRef]
- Wilson, J.A.; Hopkins, S.A.; Wright, P.M.; Dove, A.P. Synthesis of ω-Pentadecalactone Copolymers with Independently Tunable Thermal and Degradation Behavior. Macromolecules 2015, 48, 950–958. [Google Scholar] [CrossRef]
- Nifant’ev, I.E.; Shlyakhtin, A.V.; Tavtorkin, A.N.; Ivchenko, P.V.; Borisov, R.S.; Churakov, A.V. Monomeric and dimeric magnesium mono-BHT complexes as effective ROP catalysts. Catal. Commun. 2016, 87, 106–111. [Google Scholar] [CrossRef]
- Minyaev, M.E.; Churakov, A.V.; Nifant’ev, I.E. Structural diversity of polynuclear MgxOy cores in magnesium phenoxide complexes. Acta Cryst. 2017, C73, 854–861. [Google Scholar] [CrossRef]
- Minyaev, M.E.; Nifant’ev, I.E.; Shlyakhtin, A.V.; Ivchenko, P.V.; Lyssenko, K.A. Phenoxide and alkoxide complexes of Mg, Al and Zn and their use for ring-opening polymerization of ε-caprolactone with initiators of different nature. Acta Cryst. 2018, C74, 548–557. [Google Scholar] [CrossRef]
- Akatsuka, M.; Aida, T.; Inoue, S. Alcohol/methylaluminum diphenolate systems as novel, versatile initiators for synthesis of narrow molecular weight distribution polyester and polycarbonate. Macromolecules 1995, 28, 1320–1322. [Google Scholar] [CrossRef]
- Descour, C.; Sciarone, T.J.J.; Cavallo, D.; Macko, T.; Kelchtermans, M.; Korobkov, I.; Duchateau, R. Exploration of the effect of 2,6-(t-Bu)2-4-Me-C6H2OH (BHT) in chain shuttling polymerization. Polym. Chem. 2013, 4, 4718–4729. [Google Scholar] [CrossRef]
- Lozhkin, B.; Shlyakhtin, A.; Bagrov, V.; Ivchenko, P.; Nifant’ev, I. Effective stereoselective approach to substituted 1,4-dioxan-2,5-diones as prospective substrates for ring-opening polymerization. Mendeleev Commun. 2018, 28, 61–63. [Google Scholar] [CrossRef]
- Nifant’ev, I.E.; Shlyakhtin, A.V.; Bagrov, V.V.; Komarov, P.D.; Kosarev, M.A.; Tavtorkin, A.N.; Minyaev, M.E.; Roznyatovsky, V.A.; Ivchenko, P.V. Controlled ring-opening polymerisation of cyclic phosphates, phosphonates and phosphoramidates catalysed by hereroleptic BHT-alkoxy magnesium complexes. Polym. Chem. 2017, 8, 6806–6816. [Google Scholar] [CrossRef]
- Xiao, C.-S.; Wang, Y.-C.; Du, J.-Z.; Chen, X.-S.; Wang, J. Kinetics and Mechanism of 2-Ethoxy-2-oxo-1,3,2-dioxaphospholane Polymerization Initiated by Stannous Octoate. Macromolecules 2006, 39, 6825–6831. [Google Scholar] [CrossRef]
- Darensbourg, D.J.; Rainey, P.; Yarbrough, J. Bis-Salicylaldiminato Complexes of Zinc. Examination of the Catalyzed Epoxide/CO2 Copolymerization. Inorg. Chem. 2001, 40, 986–993. [Google Scholar] [CrossRef]
- Bruker. APEX-III; Bruker AXS Inc.: Madison, WI, USA, 2019. [Google Scholar]
- Krause, L.; Herbst-Irmer, R.; Sheldrick, G.M.; Stalke, D. Comparison of silver and molybdenum microfocus X-ray sources for single-crystal structure determination. J. Appl. Cryst. 2015, 48, 3–10. [Google Scholar] [CrossRef] [Green Version]
- Sheldrick, G.M. SHELXT—Integrated space-group and crystal-structure determination. Acta Cryst. 2015, A71, 3–8. [Google Scholar] [CrossRef] [Green Version]
- Sheldrick, G.M. Crystal structure refinement with SHELXL. Acta Cryst. 2015, C71, 3–8. [Google Scholar] [CrossRef]
- Macrae, C.F.; Sovago, I.; Cottrell, S.J.; Galek, P.T.A.; McCabe, P.; Pidcock, E.; Platings, M.; Shields, G.P.; Stevens, J.S.; Towler, M.; et al. Mercury 4.0: From visualization to analysis, design and prediction. J. Appl. Cryst. 2020, 53, 226–235. [Google Scholar] [CrossRef] [Green Version]
- CCDC Access Structures. Available online: https://www.ccdc.cam.ac.uk/structures/ (accessed on 1 October 2020).
- Laikov, D.N.; Ustynyuk, Y.A. PRIRODA-04: A quantum-chemical program suite. New possibilities in the study of molecular systems with the application of parallel computing. Russ. Chem. Bull. 2005, 54, 820–826. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 09, Revision, A.01; Gaussian, Inc.: Wallingford, CT, USA, 2016. [Google Scholar]
- Perdew, J.P.; Wang, Y. Accurate and Simple Analytic Representation of the Electron-Gas Correlation Energy. Phys. Rev. B 1992, 45, 13244–13249. [Google Scholar] [CrossRef] [PubMed]
- Becke, A.D. Density-functional thermochemistry. III. The role of exact exchange. J. Chem. Phys. 1993, 98, 5648–5652. [Google Scholar] [CrossRef] [Green Version]
- Sosa, C.; Andzelm, J.; Elkin, B.C.; Wimmer, E.; Dobbs, K.D.; Dixon, D.A. A local density functional study of the structure and vibrational frequencies of molecular transition-metal compounds. J. Phys. Chem. 1992, 96, 6630–6636. [Google Scholar] [CrossRef]
- Godbout, N.; Salahub, D.R.; Andzelm, J.; Wimmer, E. Optimization of Gaussian-type basis sets for local spin density functional calculations. Part I. Boron through neon, optimization technique and validation. Can. J. Chem. 1992, 70, 560–571. [Google Scholar] [CrossRef] [Green Version]
- Nifant’ev, I.; Shlyakhtin, A.; Kosarev, M.; Karchevsky, S.; Ivchenko, P. Mechanistic Insights of BHT-Mg-Catalyzed Ethylene Phosphate’s Coordination Ring-Opening Polymerization: DFT Modeling and Experimental Data. Polymers 2018, 10, 1105. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nifant’ev, I.E.; Shlyakhtin, A.V.; Kosarev, M.A.; Komarov, P.D.; Karchevsky, S.G.; Ivchenko, P.V. Data for theoretical study of the mechanisms of ring-opening polymerization of methyl ethylene phosphate. Data Brief 2019, 26, 104431. [Google Scholar] [CrossRef]
- Wang, Y.-C.; Yuan, Y.-Y.; Du, J.-Z.; Yang, X.-Z.; Wang, J. Recent progress in polyphosphoesters: From controlled synthesis to biomedical applications. Macromol. Biosci. 2009, 9, 1154–1164. [Google Scholar] [CrossRef]
- Steinbach, T.; Wurm, F.R. Poly(phosphoester)s: A new platform for degradable polymers. Angew. Chem. Int. Ed. 2015, 54, 6098–6108. [Google Scholar] [CrossRef]
- Yilmaz, Z.E.; Jérôme, C. Polyphosphoesters: New trends in synthesis and drug delivery applications. Macromol. Biosci. 2016, 16, 1745–1761. [Google Scholar] [CrossRef]
- Bauer, K.N.; Tee, H.T.; Velencoso, M.M.; Wurm, F.R. Main-chain poly(phosphoester)s: History, syntheses, degradation, bio-and flame-retardant applications. Prog. Polym. Sci. 2017, 73, 61–122. [Google Scholar] [CrossRef]
- Appukutti, N.; Serpell, C.J. High definition polyphosphoesters: Between nucleic acids and plastics. Polym. Chem. 2018, 9, 2210–2226. [Google Scholar] [CrossRef]













{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Entry | Precat. | Monomer | T, °C | Conv., % | Mn (theo), ×103 Da 1 | Mn (SEC), ×103 Da 2 | ĐM2 |
|---|---|---|---|---|---|---|---|
| 1 | 1 | lLA | 20 | >99 | 14.52 | 12.52 | 1.97 |
| 2 | 2 | lLA | 20 | 0 | - | - | - |
| 3 | 3 | lLA | 20 | 5 | - | - | - |
| 4 | 1 | lLA | 100 | >99 | 14.52 | 13.32 | 1.85 |
| 5 | 2 | lLA | 100 | 21 | - | - | - |
| 6 | 3 | lLA | 100 | >99 | 14.52 | 12.54 | 2.03 |
| 7 | 1 | εCL | 20 | >99 | 11.41 | 13.50 | 1.40 |
| 8 | 2 | εCL | 20 | >99 | 11.41 | 24.29 | 1.49 |
| 9 | 3 | εCL | 20 | 0 | - | - | - |
| 10 | 1 | εCL | 100 | >99 | 11.41 | 6.92 | 1.50 |
| 11 | 2 | εCL | 100 | 94 | 10.73 | 8.11 | 1.55 |
| 12 | 3 | εCL | 100 | 99 | 11.30 | 8.30 | 2.19 |
| Entry | Pre-Cat. | lLA/εCL/ Cat/BnOH Ratio | [lLA], mol/L | [εCL], mol/L | T, °C | t, h | Conversion, % 1 | Mn (theo), ×103 Da 2 | Mn (SEC), ×103 Da 3 | ĐM3 | CLC, % 4 | ASLC 5 | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| lLA | εCL | ||||||||||||
| 1 | 1 | 50/50/1/1 | 0.5 | 0.5 | 20 | 2 | >99 | 0 | 7.10 | 8.30 | 1.57 | 0 | - |
| 2 | 2 | 50/50/1/1 | 0.5 | 0.5 | 20 | 2 | 0 | 0 | - | - | - | 0 | - |
| 3 | 3 | 50/50/1/1 | 0.5 | 0.5 | 20 | 2 | ~5 | 0 | - | - | - | 0 | - |
| 4 | 1 | 50/50/1/1 | 0.5 | 0.5 | 100 | 2 | >99 | 0 | 7.10 | 9.70 | 1.48 | 0 | - |
| 5 | 2 | 50/50/1/1 | 0.5 | 0.5 | 100 | 2 | 25 | 0 | - | - | - | 0 | - |
| 6 | 3 | 50/50/1/1 | 0.5 | 0.5 | 100 | 2 | 99 | 36 | 8.27 | 13.15 | 1.45 | 0 | ~1.0 |
| 7a | 1 | 50/250/1/1 | 0.5 | 2.5 | 100 | 1 | 83 | 0 | 6.09 | 5.77 | 1.28 | 0 | - |
| 7b | 3 | 86 | 0 | 6.31 | 5.46 | 1.44 | 0 | - | |||||
| 7c | 5 | 87 | 0 | 6.38 | 7.30 | 1.35 | 0 | - | |||||
| 7d | 15 | 89 | 3 | 6.52 | 8.58 | 1.34 | <1 | 1.4 | |||||
| 8a | 2 | 50/250/1/1 | 0.5 | 2.5 | 100 | 1 | 30 | 3 | 3.13 | 3.39 | 1.13 | 0 | - |
| 8b | 3 | 78 | 17 | 10.58 | 9.00 | 1.15 | <1 | 3.2 | |||||
| 8c | 5 | >99 | 79 | 29.86 | 19.76 | 1.57 | 10 | 7.1 | |||||
| 8d | 15 | >99 | 98 | 35.28 | 31.90 | 1.94 | 14 | 6.8 | |||||
| 9a | 3 | 50/250/1/1 | 0.5 | 2.5 | 100 | 1 | 99 | 22 | 13.52 | 11.97 | 1.57 | 11 | 1.7 |
| 9b | 3 | >99 | 58 | 23.86 | 13.78 | 2.06 | 21 | 3.3 | |||||
| 9c | 5 | >99 | 82 | 30.71 | 20.47 | 1.72 | 31 | 4.0 | |||||
| 9d | 15 | >99 | 96 | 34.71 | 26.07 | 1.77 | 32 | 4.6 | |||||
| 10a | 4 | 50/250/1/1 | 0.5 | 2.5 | 100 | 1 | 94 | 3 | 7.74 | 13.07 | 1.26 | 0 | - |
| 10b | 3 | >99 | 43 | 19.51 | 21.73 | 1.65 | 7 | 5.7 | |||||
| 10c | 5 | >99 | 78 | 29.57 | 35.40 | 1.72 | 11 | 6.4 | |||||
| Run | Comonomer | Reaction Time, h | Conversion, % | Mn (theo), ×103 Da 1 | Mn (SEC), ×103 Da 2 | ĐM2 | ASLC 3 | |
|---|---|---|---|---|---|---|---|---|
| Comon. | εCL | |||||||
| 1 | PLLA | 5 | - | 97 | 34.99 | 22.96 | 1.74 | 6.0 |
| 2 | 15 | - | 99 | 35.56 | 27.25 | 1.85 | 5.9 | |
| 3a | MeGL | 1 | 94 | ~1 | 6.51 | 5.59 | 1.37 | ~1 |
| 3b | 3 | >99 | 58 | 23.16 | 14.25 | 1.86 | 3.1 | |
| 3c | 5 | >99 | 91 | 32.58 | 23.59 | 1.79 | 3.6 | |
| 4a | PhGL | 1 | >99 | 2 | 10.29 | 1.54 | 3.20 | - |
| 4b | 3 | >99 | 5 | 11.14 | 1.49 | 3.56 | - | |
| 4c | 5 | >99 | 12 | 13.14 | 1.51 | 5.64 | - | |
| 5a | EtEP | 1 | >99 | >99 | 35.16 | 23.02 | 1.48 | 20 |
| 5b | 3 | >99 | >99 | 35.16 | 19.23 | 1.94 | 19 | |
| 5c | 5 | >99 | >99 | 35.16 | 24.01 | 1.60 | 14.5 | |
| 6a | EtOEP | 1 | >99 | 81 | 30.83 | 36.12 | 1.91 | 6.8 |
| 6b | 3 | >99 | 82 | 31.11 | 34.55 | 2.12 | 5.2 | |
| 6c | 5 | >99 | 84 | 31.68 | 38.77 | 2.42 | 4.5 | |
| Bond | Distance, Å |
|---|---|
| Zn-O1 | 1.9265(10) |
| Zn-C28 | 1.9733(15) |
| Zn1-N1 | 2.0591(13) |
| Zn1-O2 | 2.2274(12) |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nifant’ev, I.; Komarov, P.; Ovchinnikova, V.; Kiselev, A.; Minyaev, M.; Ivchenko, P. Comparative Experimental and Theoretical Study of Mg, Al and Zn Aryloxy Complexes in Copolymerization of Cyclic Esters: The Role of the Metal Coordination in Formation of Random Copolymers. Polymers 2020, 12, 2273. https://doi.org/10.3390/polym12102273
Nifant’ev I, Komarov P, Ovchinnikova V, Kiselev A, Minyaev M, Ivchenko P. Comparative Experimental and Theoretical Study of Mg, Al and Zn Aryloxy Complexes in Copolymerization of Cyclic Esters: The Role of the Metal Coordination in Formation of Random Copolymers. Polymers. 2020; 12(10):2273. https://doi.org/10.3390/polym12102273
Chicago/Turabian StyleNifant’ev, Ilya, Pavel Komarov, Valeriya Ovchinnikova, Artem Kiselev, Mikhail Minyaev, and Pavel Ivchenko. 2020. "Comparative Experimental and Theoretical Study of Mg, Al and Zn Aryloxy Complexes in Copolymerization of Cyclic Esters: The Role of the Metal Coordination in Formation of Random Copolymers" Polymers 12, no. 10: 2273. https://doi.org/10.3390/polym12102273