Development of Novel Galactosylated PLGA Nanoparticles for Hepatocyte Targeting Using Molecular Modelling

 ,
,  , and
, and 
Abstract
:
1. Introduction
2. Materials and Methods
2.1. Molecular Modelling
2.2. Materials
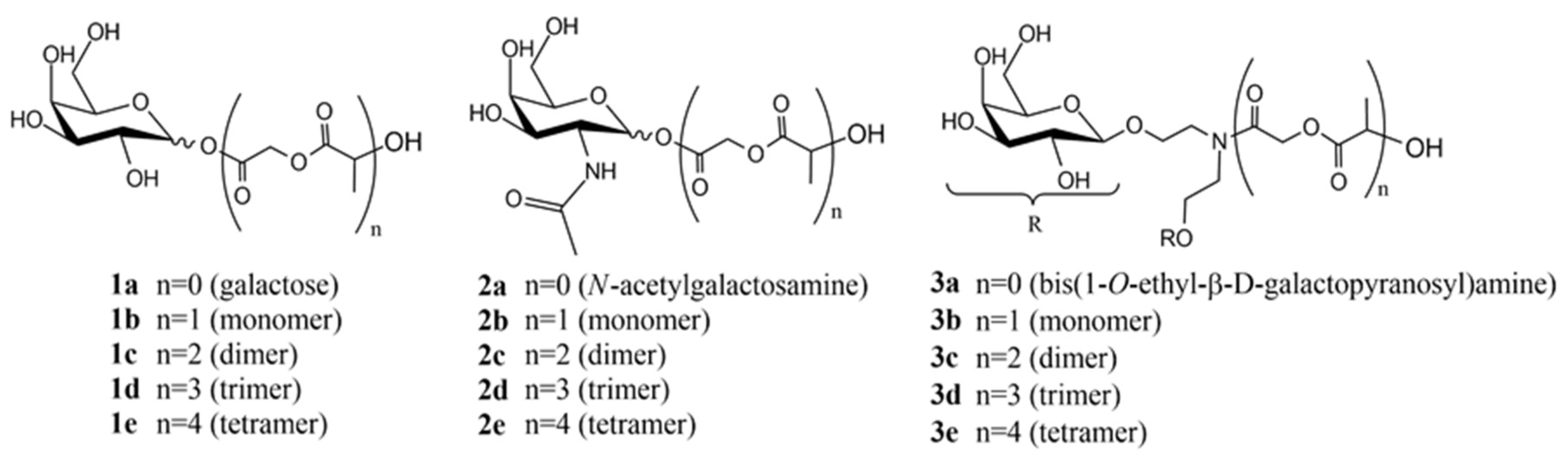
2.3. Ligand and Polymer Synthesis
2.4. Production of PLGA and PLGA-di-GAL Nanoparticles
2.5. Characterization of Nanoparticles
2.5.1. Surface Morphology
2.5.2. Particles Size and Zeta Potential
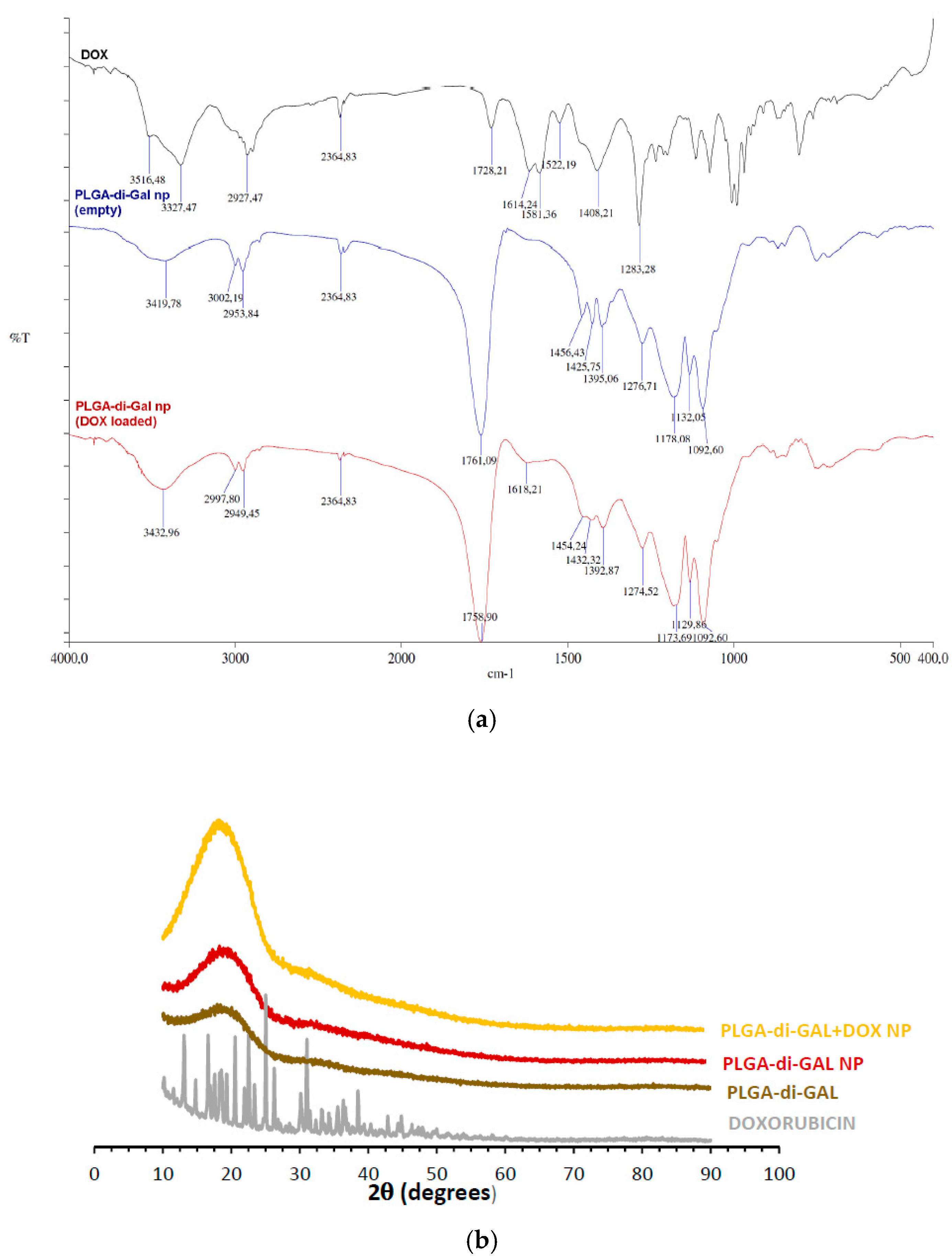
2.5.3. Fourier Transformed Infrared Spectroscopy (FTIR)
2.5.4. X-ray Diffraction (XRD)
2.5.5. Thermal Analysis
2.5.6. Drug Encapsulation Efficiency and Load Content
2.5.7. In Vitro Doxorubicin Release Studies
2.5.8. In Vitro Interaction of Galactose Conjugated NP with HepG2/C3A and HEK293T
Cell Culture
Cell Viability Assays
3. Results and Discussion
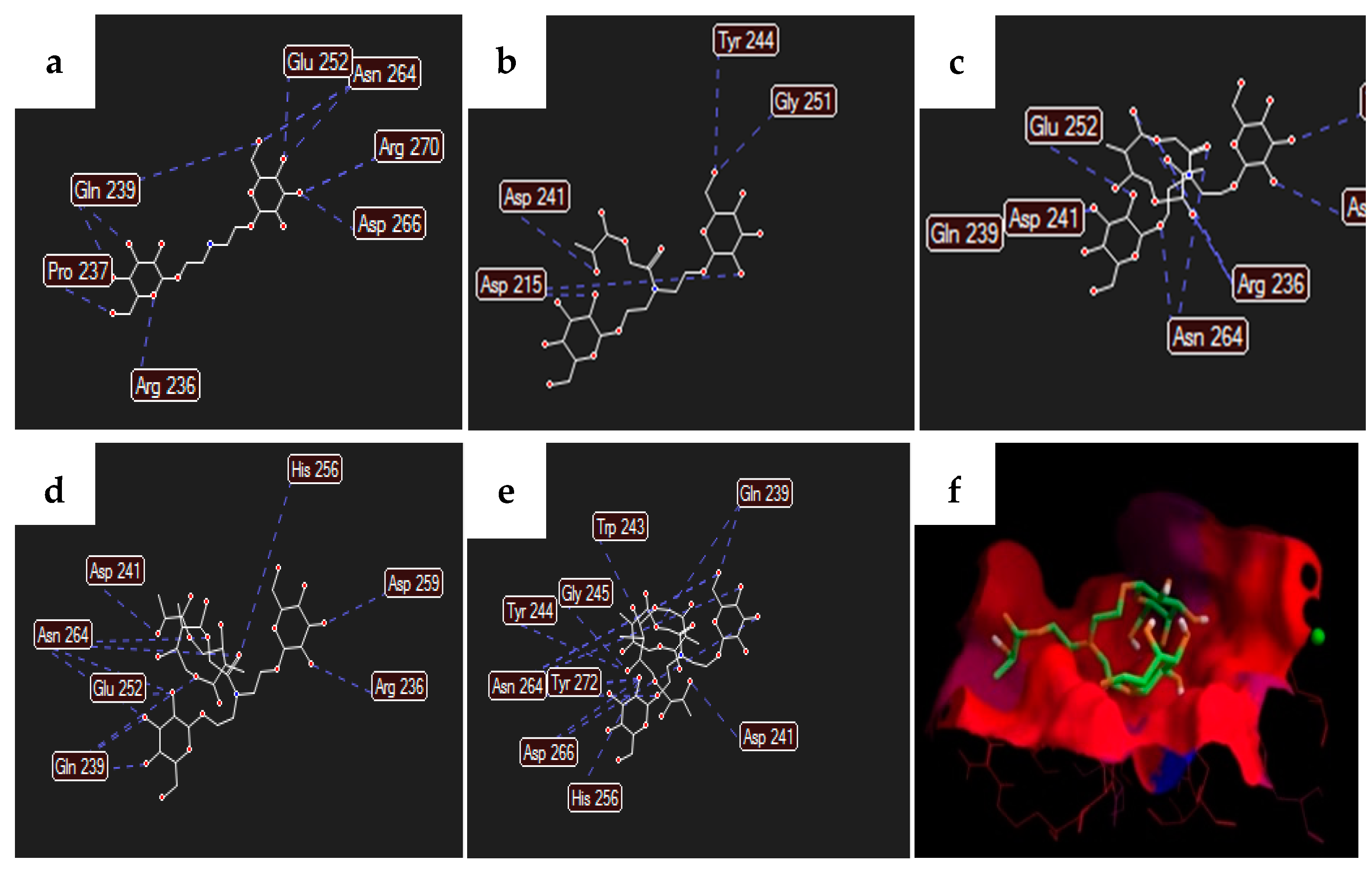
3.1. Molecular Modelling
3.2. Synthesis and Characterization of Ligand 3a and Ligand 3a-Conjugated PLGA (10)
3.3. Nanoparticle Preparation and Characterization
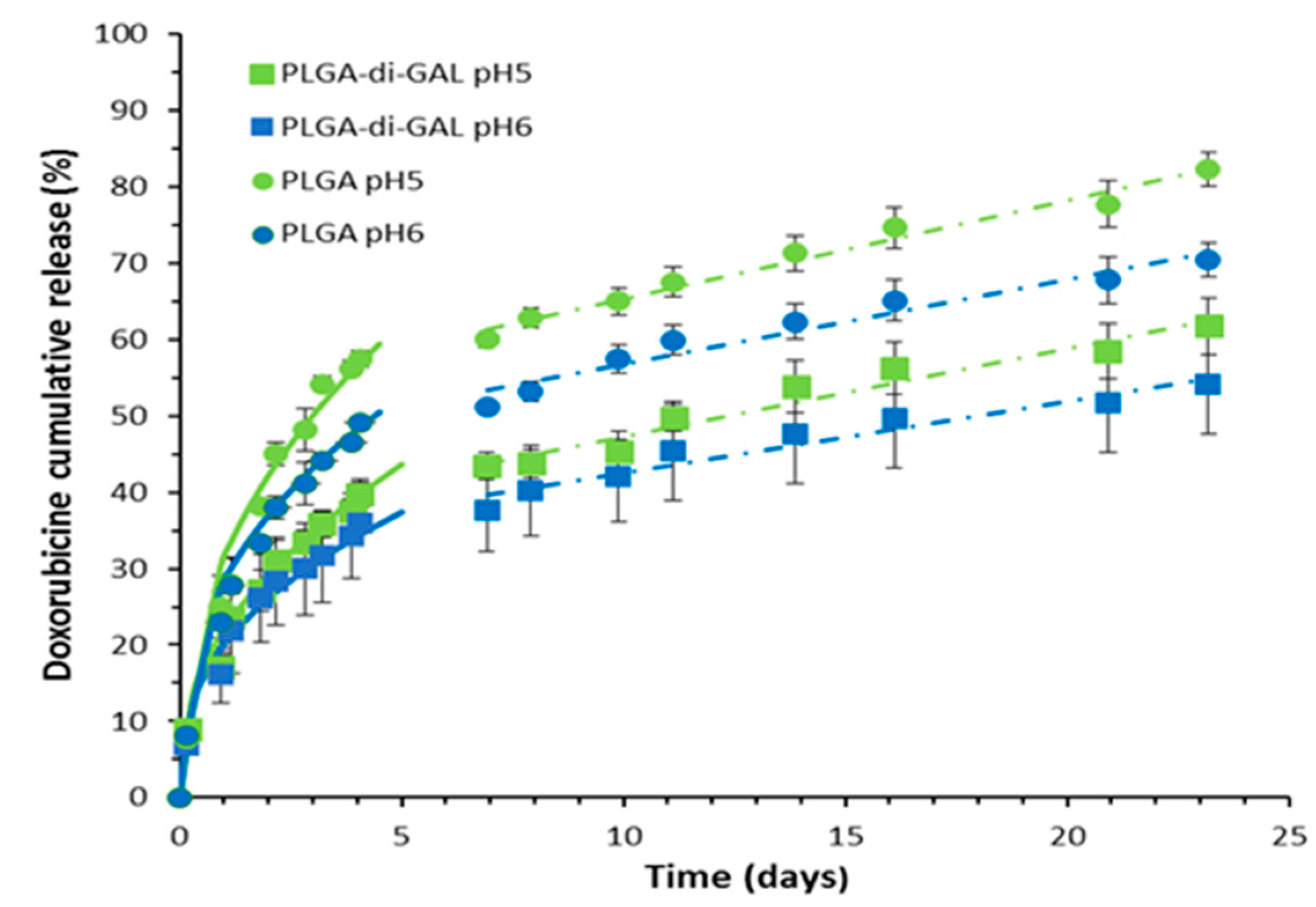
3.4. In Vitro Doxorubicin Release Studies
3.5. In Vitro Cellular Interactions of Galactose Conjugated NP with HepG2/C3A and HEK293T
3.6. In Vitro Cytotoxicity
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Merisko-Liversidge, E.; Liversidge, G.; Cooper, E. Nanosizing: A formulation approach for poorly-water-soluble compounds. Eur. J. Pharm. Sci. 2003, 18, 113–120. [Google Scholar] [CrossRef]
- Sahoo, S.K.; Labhasetwar, V. Nanotech approaches to drug delivery and imaging. Drug Discov. Today 2003, 24, 1112–1120. [Google Scholar] [CrossRef]
- Gu, F.X.; Karnik, R.; Wang, A.Z.; Alexis, F.; Levy-Nissenbaumb, E.; Hong, S.; Langer, R.S.; Farokhzad, O.C. Targeted nanoparticles for cancer therapy. Nano Today 2007, 2, 14–21. [Google Scholar] [CrossRef]
- Swainson, S.M.E.; Styliari, I.D.; Taresco, V.; Garnett, M.C. Poly(glycerol adipate) (PGA), an enzymatically synthesized functionalizable polyester and versatile drug delivery carrier: A literature update. Polymers 2019, 11, 1561. [Google Scholar] [CrossRef] [Green Version]
- Muriel, P.; Rivera-Espinoza, Y. Beneficial drugs for liver diseases. J. Appl. Toxicol. 2008, 28, 93–103. [Google Scholar] [CrossRef]
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics. CA Cancer J. Clin. 2015, 65, 5–29. [Google Scholar] [CrossRef]
- Meier, M.; Bider, M.D.; Malashkevich, V.N.; Spiess, M.; Burkhard, P. Crystal structure of the carbohydrate recognition domain of the H1 subunit of the asialoglycoprotein receptor. J. Mol. Biol. 2000, 300, 857–865. [Google Scholar] [CrossRef] [PubMed]
- Wu, G.Y.; Wu, C.H. Receptor-mediated delivery of foreign genes to hepatocytes. Adv. Drug Deliv. Rev. 1998, 29, 243–248. [Google Scholar] [CrossRef]
- Wu, J.; Sun, T.M.; Yang, X.Z.; Zhu, J.; Du, X.-J.; Yao, Y.-D.; Xiong, M.-H.; Wang, H.-X.; Wang, Y.-C.; Wang, J. Enhanced drug delivery to hepatocellular carcinoma with a galactosylated core–shell polyphosphoester nanogel. Biomater. Sci. 2013, 1, 1143–1150. [Google Scholar] [CrossRef]
- Lian, J.; Zhang, S.; Wang, J.; Fang, K.; Zhang, Y.; Hao, Y. Novel galactosylated SLN for hepatocyte-selective targeting of floxuridinyl diacetate. J. Drug Target. 2008, 16, 250–256. [Google Scholar] [CrossRef]
- Khorev, O.; Stokmaier, D.; Schwardt, O. Trivalent, Gal/GalNAc-containing ligands designed for the asialoglycoprotein receptor. Bioorg. Med. Chem. 2008, 16, 5216–5231. [Google Scholar] [CrossRef] [PubMed]
- Guo, B.; Cheng, Y.; Li, N.; Li, X.; Jin, M.; Li, T.; Li, J. In vitro and in vivo studies of galactose-modified liver-targeting liposomes. J. Drug Target. 2012, 21, 257–264. [Google Scholar] [CrossRef] [PubMed]
- Cardoso, M.M.; Peca, I.N.; Raposo, C.D.; Petrova, K.T.; Barros, M.T.; Gardner, R.; Bicho, A. Doxorubicin-loaded galactose-conjugated poly(d,l-lactide-co-glycolide) nanoparticles as hepatocyte-targeting drug carrier. J. Microencapsul. 2016, 33, 315–322. [Google Scholar] [CrossRef] [PubMed]
- Hoffman, A.S. Selecting the right polymer for biomaterial applications. In Polymers in Drug Delivery; Uchegbu, I.F., Schätzlein, A.G., Eds.; CRC/Taylor & Francis: Boca Raton, FL, USA, 2006. [Google Scholar]
- Arcamone, F. Doxorubicin: Anticancer Antibiotics; Academic Press: New York, NY, USA, 1981. [Google Scholar]
- Monneret, C. Recent developments in the field of antitumour anthracyclines. Eur. J. Med. Chem. 2001, 36, 483–493. [Google Scholar] [CrossRef]
- Trott, O.; Olson, A. AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization and multithreading. J. Comput. Chem. 2010, 31, 455–461. [Google Scholar] [CrossRef] [Green Version]
- Baenziger, J.U.; Fiete, D. Galactose and N-acetylgalactosamine-specific endocytosis of glycopeptides by isolated rat hepatocytes. Cell 1980, 22, 611. [Google Scholar] [CrossRef]
- Perrin, D.D.; Armarego, W.L.F. Purification of Laboratory Chemicals; Butterworth-Heinemann: Oxford, UK, 1988. [Google Scholar]
- Shendage, D.M.; Fröhlich, R.; Haufe, G. Highly efficient stereoconservative amidation and deamidation of α-amino acids. Org. Lett. 2004, 6, 3675–3678. [Google Scholar] [CrossRef]
- Kolatkar, A.R.; Leung, A.K.; Isecke, R.; Brossmer, R.; Drickamer, K.; Weis, W. Mechanism of N-acetylgalactosamine binding to a C-type animal lectin carbohydrate-recognition domain. J. Biol. Chem. 1998, 273, 19502–19508. [Google Scholar] [CrossRef] [Green Version]
- Bergmeier, S.C.; Fundy, S.L. Synthesis of oligo(5-aminopentanoic acid)-nucleobases (APN): Potential antisense agents. Bioorg. Med. Chem. Lett. 1997, 7, 3135–3138. [Google Scholar] [CrossRef]
- Goldschmid, H.R.; Perlin, A.S. Some factors affecting the Königs-Knorr synthesis of glycosides. Can. J. Chem. 1961, 39, 2025–2034. [Google Scholar] [CrossRef] [Green Version]
- Kimmel, R.; Kafka, S.; Kosmrlj, J. Selective formation of glycosidic linkages of N-unsubstituted 4-hydroxyquinolin-2-(1H)-ones. Carbohyd. Res. 2010, 345, 768–779. [Google Scholar] [CrossRef] [PubMed]
- Stick, R.V.; Williams, S.J. Carbohydrates: The Essential Molecules of Life; Elsevier: Amsterdam, The Netherlands, 2009. [Google Scholar]
- Peca, I.N.; Petrova, K.T.; Cardoso, M.M.; Barros, M.T. Preparation and characterization of polymeric nanoparticles composed of poly(dl-lactide-co-glycolide) and poly(dl-lactide-co-glycolide)-co-poly(ethylene glycol)-10%-Triblock end-capped with a galactose moiety. React. Funct. Polym. 2012, 72, 729–735. [Google Scholar] [CrossRef]
- Ward, C.M.; Read, M.L.; Seymour, L.W. Systemic circulation of poly(l-lysine)/DNA vectors is influenced by polycation molecular weight and type of DNA: Differential circulation in mice and rats and the implications for human gene delivery. Blood 2001, 97, 2221–2229. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kashiwagura, T.; Deutsch, C.J.; Taylor, J.; Wilson, D.F. Dependence of gluconeogenesis, urea synthesis and energy metabolism of hepatocytes on intracellular pH. J. Biol. Chem. 1984, 259, 237–243. [Google Scholar] [PubMed]
- Gores, G.J.; Nieminen, A.L.; Wray, B.E. Intracellular pH during “chemical hypoxia” in cultured rat hepatocytes. Protection by intracellular acidosis against the onset of cell death. J. Clin. Invest. 1989, 83, 386–396. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kharbanda, K.K.; McVicker, D.L.; Zetterman, R.K.; MacDonald, R.G.; Donohue, T.M., Jr. Flow cytometric analysis of vesicular pH in rat hepatocytes after ethanol administration. Hepatology 1997, 26, 929–934. [Google Scholar] [CrossRef]
- Kim, H.; Fassihi, R. Application of binary polymer system in drug release rate modulation. 2. Influence of formulation variables and hydrodynamic conditions on release kinetics. J. Pharm. Sci. 1997, 83, 323–328. [Google Scholar] [CrossRef]
- Ford, J.L.; Mitchell, K.; Rowe, P.; Armstrong, D.J.; Elliott, P.N.C.; Rostron, C.; Hogan, J.E. Mathematical modelling of drug release from hydroxypropylmethylcellulose matrices: Effect of temperature. Int. J. Pharm. 1997, 71, 95–104. [Google Scholar] [CrossRef]
- Ritger, P.L.; Peppas, N.A. A simple equation for description of solute release II. Fickian and anomalous release from swellable devices. J. Control. Release 1987, 5, 37–42. [Google Scholar] [CrossRef]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | Binding Energy (kcal/mol) | ||
|---|---|---|---|
| N = 1 (Galactose) | N = 2 (N-acetylgalactosamine) | N = 3 (bis(1-O-ethyl-β-D-galactopyranosyl)amine) | |
| Na | −5.3 | −5.4 | −6.0 |
| Nb | −6.0 | −6.7 | −6.5 |
| Nc | −6.0 | −6.8 | −6.1 |
| Nd | −5.3 | −6.2 | −5.9 |
| Ne | −5.5 | −5.6 | −5.9 |
| System | Particle Size (nm) a | Zeta Potential (mV) a | Tg (°C) b | EE (% w/w) c | DOX Content (% w/w) d |
|---|---|---|---|---|---|
| PLGA | 43.32 | ||||
| PLGA-di-GAL | -- | -- | 44.47 | -- | -- |
| PLGA NP | 238 ± 48 | −18.4 | 45.55 | -- | -- |
| PLGA-di-GAL NP | 311 ± 58 | −30.3 | 44.14 | -- | -- |
| DOX-PLGA NP | 239 ± 43 | −71.7 | 44.9 | 65 ± 10 | 2.9 ± 0.4 |
| DOX-PLGA-di-GAL NP | 258 ± 47 | −62.3 | 45.69 | 83 ± 9 | 3.7 ± 0.4 |
| Particle | pH | n | n Error (%) | k (day−1) | k Error (%) | r2 |
|---|---|---|---|---|---|---|
| PLGA-di-GAL NP | pH5 | 0.494 | 5.4 | 14.6 | 6.2 | 0.99 |
| pH6 | 0.474 | 4.4 | 14.5 | 6.3 | 0.99 | |
| PLGA NP | pH5 | 0.450 | 13.1 | 25.0 | 8.3 | 0.99 |
| pH6 | 0.446 | 3.83 | 19.2 | 3.0 | 0.98 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Raposo, C.D.; Costa, R.; Petrova, K.T.; Brito, C.; Scotti, M.T.; Cardoso, M.M. Development of Novel Galactosylated PLGA Nanoparticles for Hepatocyte Targeting Using Molecular Modelling. Polymers 2020, 12, 94. https://doi.org/10.3390/polym12010094
Raposo CD, Costa R, Petrova KT, Brito C, Scotti MT, Cardoso MM. Development of Novel Galactosylated PLGA Nanoparticles for Hepatocyte Targeting Using Molecular Modelling. Polymers. 2020; 12(1):94. https://doi.org/10.3390/polym12010094
Chicago/Turabian StyleRaposo, Cláudia D., Rita Costa, Krasimira T. Petrova, Catarina Brito, Marcus T. Scotti, and M. Margarida Cardoso. 2020. "Development of Novel Galactosylated PLGA Nanoparticles for Hepatocyte Targeting Using Molecular Modelling" Polymers 12, no. 1: 94. https://doi.org/10.3390/polym12010094