A Robust, Tough and Multifunctional Polyurethane/Tannic Acid Hydrogel Fabricated by Physical-Chemical Dual Crosslinking


Abstract
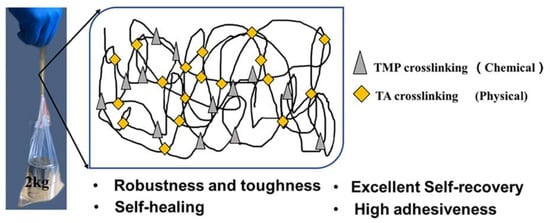
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Fabrication of Polyethylene Polyurethane (PEG–PU) Hydrogels
2.3. Fabrication of TA–PU Hydrogels
2.4. Characterizations and Measurements
2.4.1. Fourier Transform Infrared Spectroscopy (FTIR)
2.4.2. Scanning Electron Microscope (SEM)
2.4.3. X-ray Diffraction Pattern (XRD)
2.4.4. Swelling Behavior
2.4.5. Mechanical Performance
2.4.6. Dynamic Mechanical Analysis (DMA)
2.4.7. Adhesiveness Test
3. Results and Discussion
3.1. Fabrication of TA–PU Hydrogels
3.2. Structural Characterization
3.3. Microstructure Characterization
3.4. Swelling Behavior
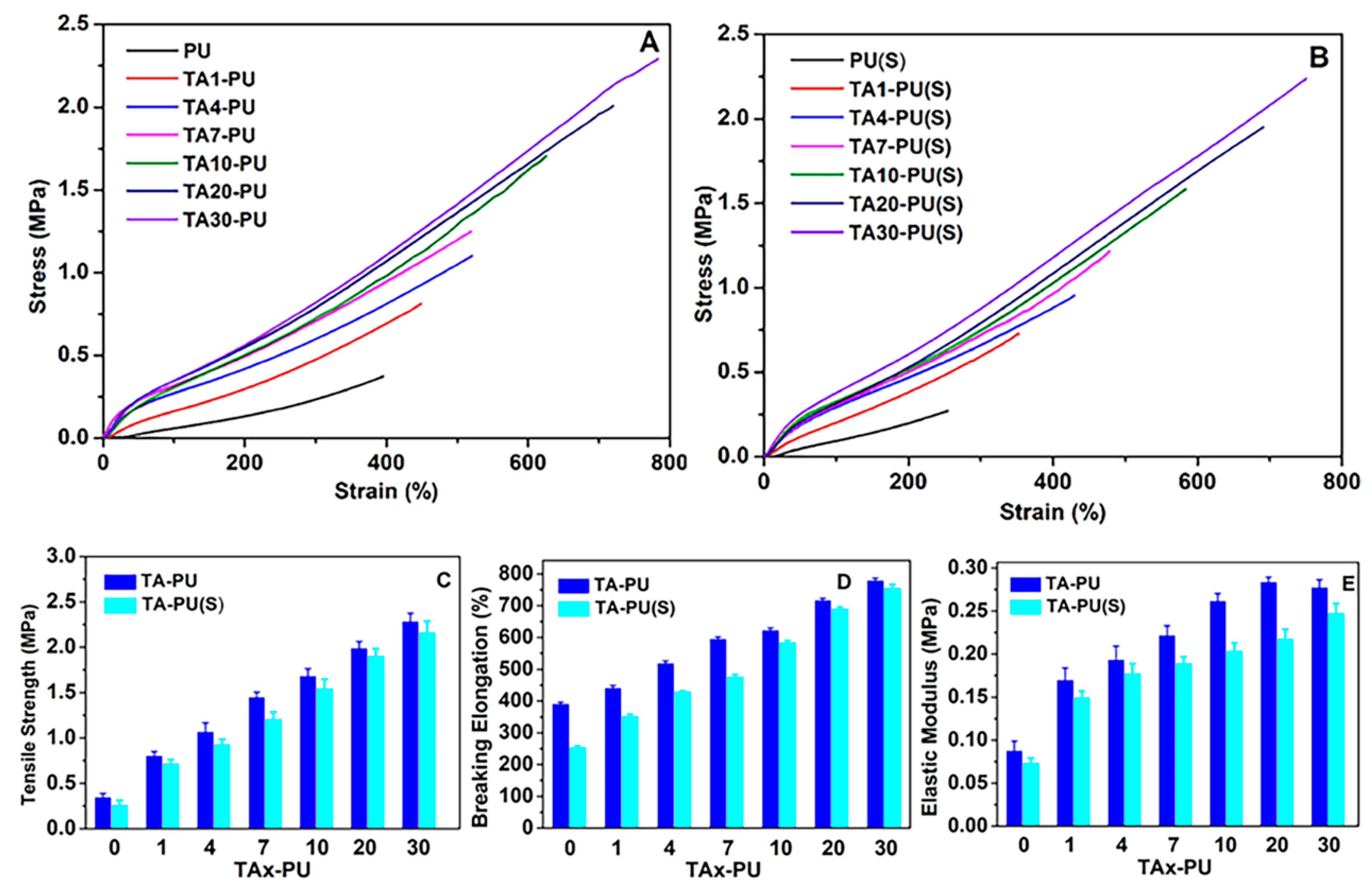
3.5. Mechanical Properties
3.6. Toughness of TA–PU Hydrogels
3.7. Dynamic Mechanical Performance of TA–PU Gels
3.8. Self-Recovery and Self-Healing Properties of TA–PU Hydrogels
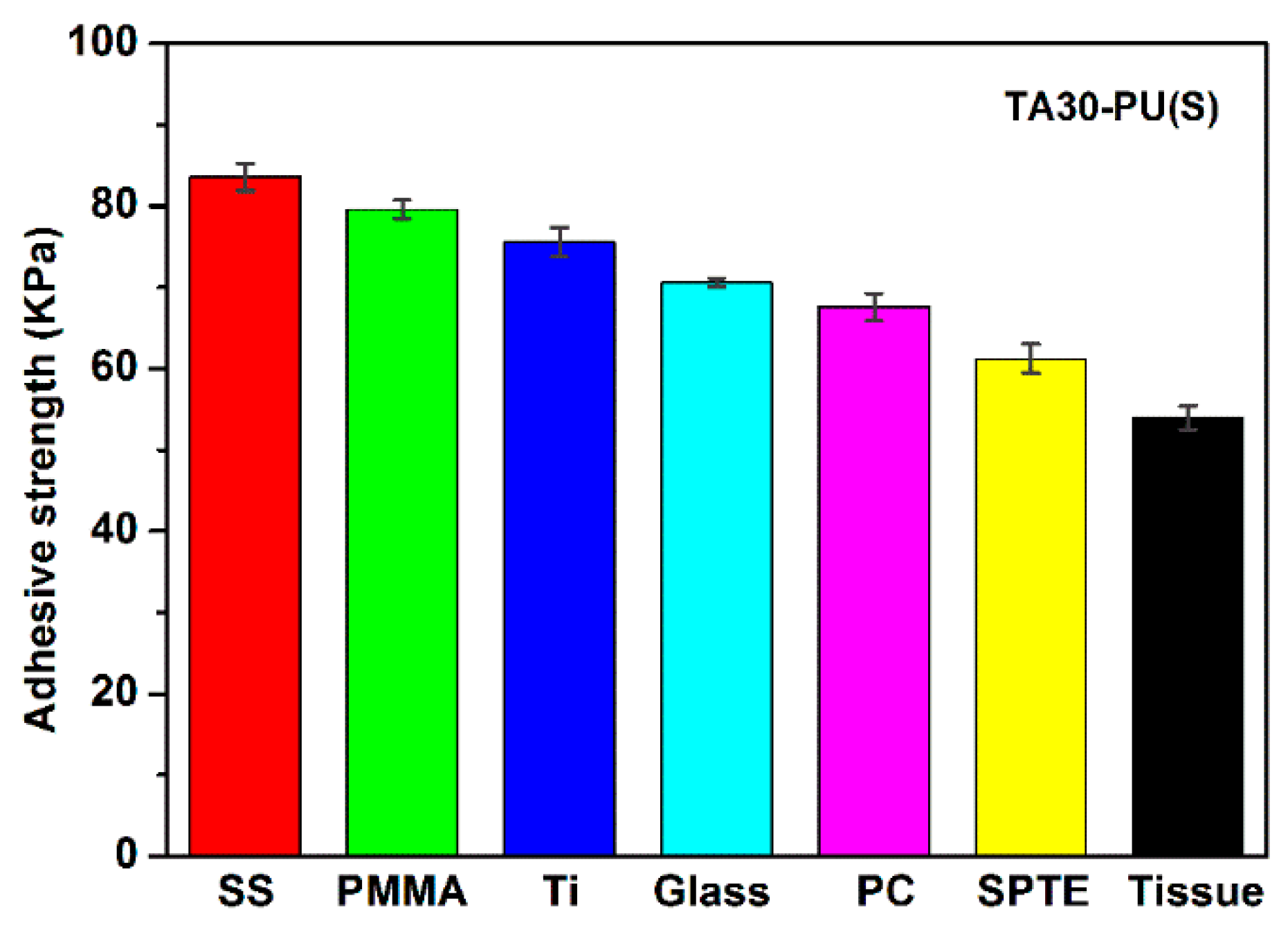
3.9. Adhesiveness of TA–PU Hydrogels
4. Conclusions
Supplementary Materials
Author Contributions
Acknowledgments
Conflicts of Interest
References
- Ahmed, E.M. Hydrogel: Preparation, characterization, and applications: A review. J. Adv. Res. 2015, 6, 105–121. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nie, J.Y.; Pei, B.Y.; Wang, Z.K.; Hu, Q.L. Construction of ordered structure in polysaccharide hydrogel: A review. Carbohydr. Polym. 2019, 205, 225–235. [Google Scholar] [CrossRef] [PubMed]
- Rosiaka, J.M.; Yoshii, F. Hydrogels and their medical applications. Nucl. Instrum. Methods B 1999, 151, 56–64. [Google Scholar] [CrossRef]
- Townsenda, J.M.; Beck, E.C.; Gehrkec, S.H.; Berklandc, C.J.; Detamore, M.S. Flow behavior prior to crosslinking: The need for precursor rheology for placement of hydrogels in medical applications and for 3D bioprinting. Prog. Polym. Sci. 2019, 91, 126–140. [Google Scholar] [CrossRef] [PubMed]
- Guilherme, M.R.; Aouadab, F.A.; Fajardoc, A.R.; Martinsa, A.F.; Paulinoe, A.T.; Davif, M.F.T.; Rubiraa, A.F.; Muniz, E.C. Superabsorbent hydrogels based on polysaccharidesfor application in agriculture as soil conditioner and nutrientcarrier: A review. Eur. Polym. J. 2015, 72, 365–385. [Google Scholar] [CrossRef] [Green Version]
- Sannino, A.; Demitri, C.; Madaghiele, M. Biodegradable cellulose-based hydrogels: Design and applications. Materials 2009, 2, 353–373. [Google Scholar] [CrossRef]
- Ullah, F.; Othman, M.B.; Javed, H.F.; Ahmad, Z.; Akil, H.M. Classification, processing and application of hydrogels: A review. Mater. Sci. Eng. C-Mater. 2015, 57, 414–433. [Google Scholar] [CrossRef]
- Sun, J.Y.; Zhao, X.; Illeperuma, W.R.; Chaudhuri, O.; Oh, K.H.; Mooney, D.J.; Vlassak, J.J.; Suo, Z. Highly stretchable and tough hydrogels. Nature 2012, 489, 133–136. [Google Scholar] [CrossRef]
- Wang, E.; Desai, M.S.; Lee, S.W. Light-controlled graphene-elastin composite hydrogel actuators. Nano Lett. 2013, 13, 2826–2830. [Google Scholar] [CrossRef] [Green Version]
- Calvert, P. Hydrogels for soft machines. Adv. Mater. 2009, 21, 743–756. [Google Scholar] [CrossRef]
- Ding, B.; Gao, H.; Song, J.; Li, Y.; Zhang, L.; Cao, X.; Xu, M.; Cai, J. Tough and cell-compatible chitosan physical hydrogels for mouse bone mesenchymal stem cells in vitro. ACS Appl. Mater. Interfaces 2016, 8, 19739–19746. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Illeperuma, W.R.K.; Suo, Z.; Vlassak, J.J. Hybrid hydrogels with extremely high stiffness and toughness. ACS Macro Lett. 2014, 3, 520–523. [Google Scholar] [CrossRef]
- Kaneko, D.; Gong, J.P.; Osada, Y. Polymer gels as soft and wet chemomechanical systems-an approach to artificial Muscles. J. Mater. Chem. 2002, 12, 2169–2177. [Google Scholar] [CrossRef]
- Peppas, N.A.; Hilt, J.Z.; Khademhosseini, A.; Langer, R. Hydrogels in biology and medicine: From molecular principles to bionanotechnology. Adv. Mater. 2006, 18, 1345–1360. [Google Scholar] [CrossRef]
- Gong, J.P.; Osada, Y. Soft and wet materials: From hydrogels to biotissues. Adv. Polym. Sci. 2010, 236, 203–246. [Google Scholar]
- Chang, C.W.; Spreeuwel, A.V.; Zhang, C.; Varghese, S. PEG/clay nanocomposite hydrogel: A mechanically robust tissue engineering scaffold. Soft Matter 2010, 6, 5157–5164. [Google Scholar] [CrossRef]
- Gong, J.P.; Katsuyama, Y.; Kurokawa, T.; Osada, Y. Double-network hydrogels with extremely high mechanical strength. Adv. Mater. 2003, 15, 1155–1158. [Google Scholar] [CrossRef]
- Fan, H.L.; Wang, J.H.; Jin, Z.X. Tough, swelling-Resistant, self-Healing, and adhesive dual-crosslinked hydrogels based on polymer—Tannic acid multiple hydrogen bonds. Macromolecules 2018, 15, 1696–1705. [Google Scholar] [CrossRef]
- Krsko, P.; Libera, M. Biointeractive hydrogels. Mater. Today 2005, 8, 36–44. [Google Scholar] [CrossRef]
- Sun, P.Y.; Wang, J.; Xiong, Y.; Peng, Y.; Peng, X.; Tu, X.; Du, P.F.; Zheng, Z.; Wang, X.L. Facile preparation of mussel-inspired polyurethane hydrogel and its rapid curing behavior. ACS Appl. Mater. Interfaces 2014, 6, 12495–12504. [Google Scholar] [CrossRef]
- Dong, Y.M.; Jin, Y.W.; Zhang, Z.J. Synthesis and application of polyurethane Hydrogel carrier on nitrobacteria immobilization. Adv. Mater. Res. 2010, 152, 1533–1536. [Google Scholar] [CrossRef]
- Corneillie, S.; Lan, P.N.; Schacht, E.; Davies, M.; Shard, A.; Green, R.; Wassall, S.M.; Whitfield, H.; Choong, S. Polyethylene glycol-containing polyurethanes for biomedical applications. Polym. Int. 1998, 46, 251–259. [Google Scholar] [CrossRef]
- Nie, T.; Baldwin, A.; Yamaguchi, N.; Kiick, K.L. Production of heparin-functionalized hydrogels for the development of responsive and controlled growth factor delivery systems. J. Control. Release 2007, 122, 287–296. [Google Scholar] [CrossRef] [Green Version]
- Leach, J.B.; Schmidt, C.E. Characterization of protein release from photocrosslinkable hyaluronic acid-polyethylene glycol hydrogel tissue engineering scaffolds. Biomaterials 2005, 26, 125–135. [Google Scholar] [CrossRef] [PubMed]
- Bell, C.L.; Peppas, N.A. Water, solute and protein diffusion in physiologically responsive hydrogels of poly methacrylic acid-g-ethylene glycol. Biomaterials 1996, 17, 1203–1218. [Google Scholar] [CrossRef]
- Lu, S.; Anseth, K.S. Release behavior of high molecular weight solutes from poly ethylene glycol-based degradable networks. Macromolecules 2000, 33, 2509–2515. [Google Scholar] [CrossRef]
- Okada, N.T.; Horiuchi, H.; Murakami, N.; Takahashi, J.; Nawata, M.; Ota, H.; Nozaki, K.; Takaoka, K. A biodegradable polymer as a cytokine delivery system for inducing bone formation. Nat. Biotechnol. 2001, 19, 332–335. [Google Scholar]
- Langer, R.; Peppas, N. Chemical and physical structure of polymers as carriers for controlled release of bioactive agents: A review. J. Macromol. Sci.-Pol. R 1983, 23, 61–126. [Google Scholar] [CrossRef]
- Li, R.; Zhou, H.H.; Li, T.; Li, C.Y.; Duan, Y.Y. Polyethylene glycol-containing polyurethane hydrogel coatings for improving the biocompatibility of neural electrodes. Acta Biomater. 2012, 8, 2233–2242. [Google Scholar]
- Hsieha, F.Y.; Lina, H.H.; Hsu, S.H. 3D bioprinting of neural stem cell-laden thermoresponsive biodegradable polyurethane hydrogel and potential in central nervous system repair. Biomaterials 2015, 71, 48–57. [Google Scholar] [CrossRef]
- Mahanta, A.K.; Sudipta, S.; Maiti, P. Polyurethane-chitosan brush as injectable hydrogel for controlled drug delivery and tissue engineering. Polym. Chem. 2017, 8, 6233–6249. [Google Scholar] [CrossRef]
- Petrini, P.; Farè, S.; Piva, A.; Tanzi, M.C. Design, synthesis and properties of polyurethane hydrogels for tissue engineering. J. Mater. Sci. 2003, 14, 683–686. [Google Scholar]
- Li, J.Z.; Ma, L.; Chen, G.X.; Zhou, Z.; Li, Q.F. High water-content and high elastic dual-responsive polyurethane hydrogel for drug delivery. J. Mater. Chem. B 2015, 3, 8401–8409. [Google Scholar] [CrossRef]
- Divakaran, A.V.; Azad, L.B.; Surwase, S.S.; Torris AT, A.; Badiger, M.V. Mechanically tunable curcumin incorporated polyurethane hydrogels as potential biomaterials. Chem. Mater. 2016, 28, 2120–2130. [Google Scholar] [CrossRef]
- Lopes, G.K.B.; Schulman, H.M.; Hermes-Lima, M. Polyphenol tannic acid inhibits hydroxyl radical formation from fenton reaction by complexing ferrous ions. BBA-Gen. Subj. 1999, 1472, 142–152. [Google Scholar] [CrossRef]
- Liao, M.H.; Wan, P.B.; Wen, J.R.; Gong, M.; Wu, X.; Wang, Y.G.; Shi, R.; Zhang, L.Q. Wearable, healable, and adhesive epidermal sensors assembled from mussel-inspired conductive hybrid hydrogel framework. Adv. Funct. Mater. 2017, 27, 1703852. [Google Scholar] [CrossRef]
- Oveissi, F.; Naficy, S.; Le, T.Y.L.; Fletcher, D.F.; Dehghani, F. Tough and processable hydrogels based on lignin and hydrophilic polyurethane. ACS Appl. Bio Mater. 2018, 1, 2073–2081. [Google Scholar] [CrossRef]
- Zheng, H.; Pan, M.W.; Wen, J.; Yuan, J.F.; Zhu, L.; Yu, H.F. Robust, transparent, and superhydrophobic coating fabricated with waterborne polyurethane and inorganic nanoparticle composites. Ind. Eng. Chem. Res. 2019, 58, 8050–8060. [Google Scholar] [CrossRef]
- Greensmith, H.W.; Thomas, A.G. Rupture of rubber. Ш. Determination of tear properties. J. Polym. Sci. Pol. Phys. 1955, 18, 189–200. [Google Scholar]
- Gogoi, S.; Barua, S.; Karak, N. Biodegradable and thermostable synthetic hyperbranched poly(urethane-urea,s as advanced surface coating Materials. Prog. Org. Coat. 2014, 77, 1418–1427. [Google Scholar] [CrossRef]
- Gogoi, S.; Karak, N. Biobased biodegradable waterborne hyperbranched polyurethane as an ecofriendly sustainable material. ACS Sustain. Chem. Eng. 2014, 2, 2730–2738. [Google Scholar] [CrossRef]
- Li, F.J.; Zhang, S.D.; Liang, J.Z.; Wang, J.Z. Effect of polyethylene glycol on the crystallization and impact properties of polylactide-based blends. Polym. Adv. Technol. 2015, 26, 465–475. [Google Scholar] [CrossRef]
- Chen, Y.N.; Peng, L.; Liu, T.; Wang, Y.; Shi, S.; Wang, H. Poly (vinyl alcohol–tannic acid hydrogels with excellent mechanical properties and shape memory behaviors. ACS Appl. Mater. Interfaces 2016, 8, 27199–27206. [Google Scholar] [CrossRef] [PubMed]
- Sun, T.L.; Luo, F.; Kurokawa, T.; Karobi, S.N.; Nakajima, T.; Gong, J.P. Molecular structure of self-healing polyampholyte hydrogels analyzed from tensile behaviors. Soft Matter 2015, 11, 9355–9366. [Google Scholar] [CrossRef] [PubMed]
- Webber, R.E.; Creton, C.; Brown, H.R.; Gong, J.P. Large strain hysteresis and mullins effect of tough double-network hydrogels. Macromolecules 2007, 40, 2919–2927. [Google Scholar] [CrossRef]
- Naficy, S.; Brown, H.R.; Razal, J.M.; Spinks, G.M.; Whitten, P.G. Progress toward robust polymer hydrogels. Aust. J. Chem. 2011, 64, 1007–1025. [Google Scholar] [CrossRef]
- Zhang, Y.Y.; Li, Y.M.; Liu, W.G. Dipole–dipole and H-bonding interactions significantly enhance the multifaceted mechanical properties of thermoresponsive shape memory hydrogels. Adv. Funct. Mater. 2015, 25, 471–480. [Google Scholar] [CrossRef]
- Song, Y.; Liu, Y.; Qi, T.; Li, G.L. Towards dynamic but supertough healable polymers through bio-mimetic hierarchical hydrogen-bonding interactions. Angew. Chem. Int. Ed. 2018, 57, 13838–13842. [Google Scholar] [CrossRef]
- Xiao, K.C.; Wang, Z.Y.; Wu, Y.J.; Lin, W.W.; He, Y.Y.; Zhan, J.H.; Luo, F.; Li, Z.; Li, J.H.; Tan, H.; et al. Biodegradable, anti-adhesive and tough polyurethane hydrogels crosslinked by triol crosslinkers. J. Biomed. Mater. Res. 2019, 107, 2205–2221. [Google Scholar] [CrossRef]
- Fan, H.L.; Wang, J.H.; Zhang, Q.Y.; Jin, Z.X. Tannic acid-based multifunctional hydrogels with facile adjustable adhesion and cohesion contributed by polyphenol supramolecular chemistry. ACS Omega 2017, 2, 6668–6676. [Google Scholar] [CrossRef]
- Kord Forooshani, P.; Lee, B.P. Recent approaches in designing bioadhesive materials inspired by mussel adhesive protein. J. Polym. Sci. Pol. Chem. 2017, 55, 9–33. [Google Scholar] [CrossRef] [PubMed]











© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wen, J.; Zhang, X.; Pan, M.; Yuan, J.; Jia, Z.; Zhu, L. A Robust, Tough and Multifunctional Polyurethane/Tannic Acid Hydrogel Fabricated by Physical-Chemical Dual Crosslinking. Polymers 2020, 12, 239. https://doi.org/10.3390/polym12010239
Wen J, Zhang X, Pan M, Yuan J, Jia Z, Zhu L. A Robust, Tough and Multifunctional Polyurethane/Tannic Acid Hydrogel Fabricated by Physical-Chemical Dual Crosslinking. Polymers. 2020; 12(1):239. https://doi.org/10.3390/polym12010239
Chicago/Turabian StyleWen, Jie, Xiaopeng Zhang, Mingwang Pan, Jinfeng Yuan, Zhanyu Jia, and Lei Zhu. 2020. "A Robust, Tough and Multifunctional Polyurethane/Tannic Acid Hydrogel Fabricated by Physical-Chemical Dual Crosslinking" Polymers 12, no. 1: 239. https://doi.org/10.3390/polym12010239