Effect of Silane Treatment on Mechanical Properties of Polyurethane/Mesoscopic Fly Ash Composites

Abstract
:1. Introduction
2. Experiments
2.1. Components of Experimental Materials
2.2. Optimization of GPTMS Dosage
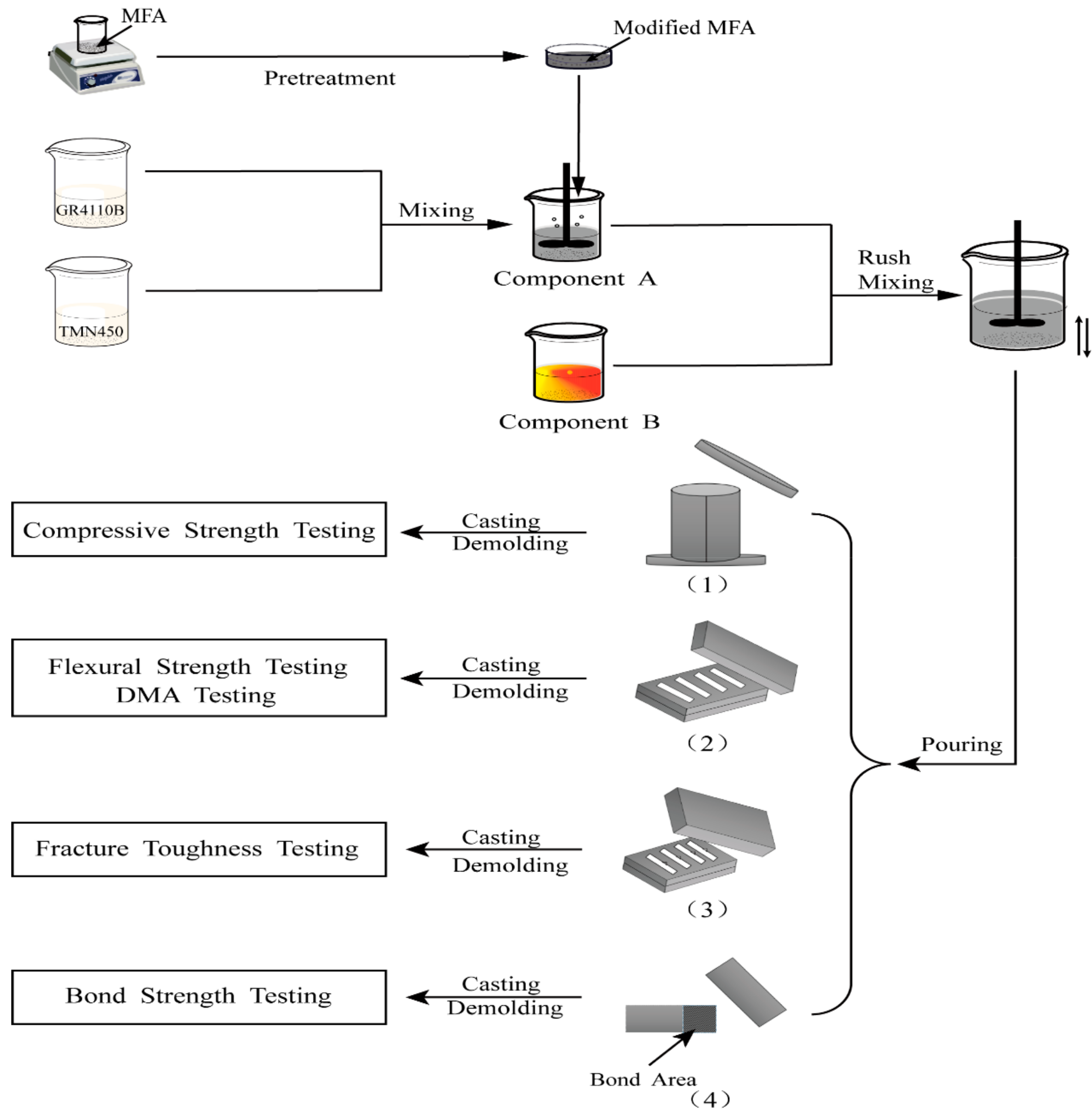
2.3. Preparation for Specimens
2.4. Analyses and Characterization Techniques
3. Results and Discussion
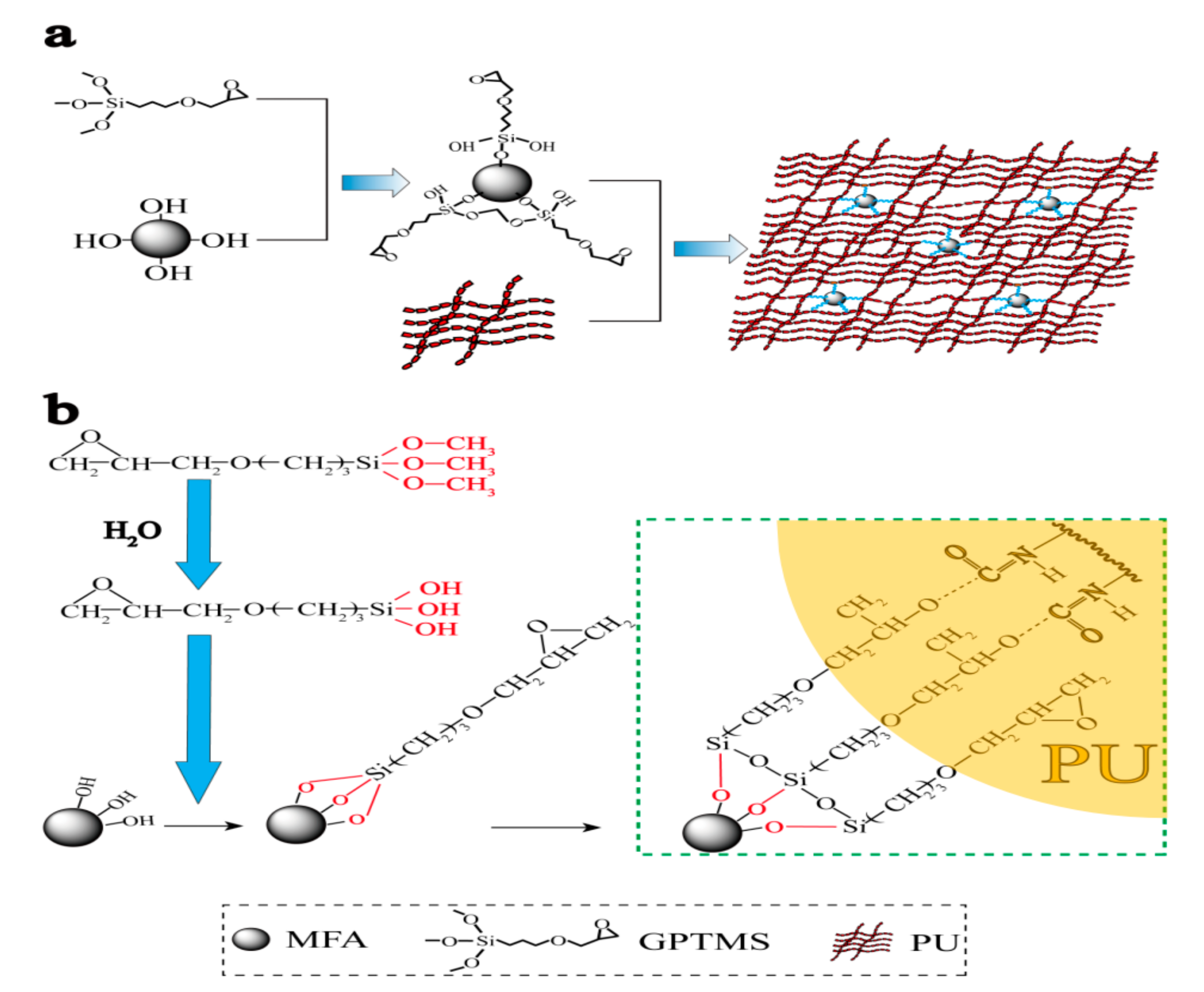
3.1. Mechanism Analysis
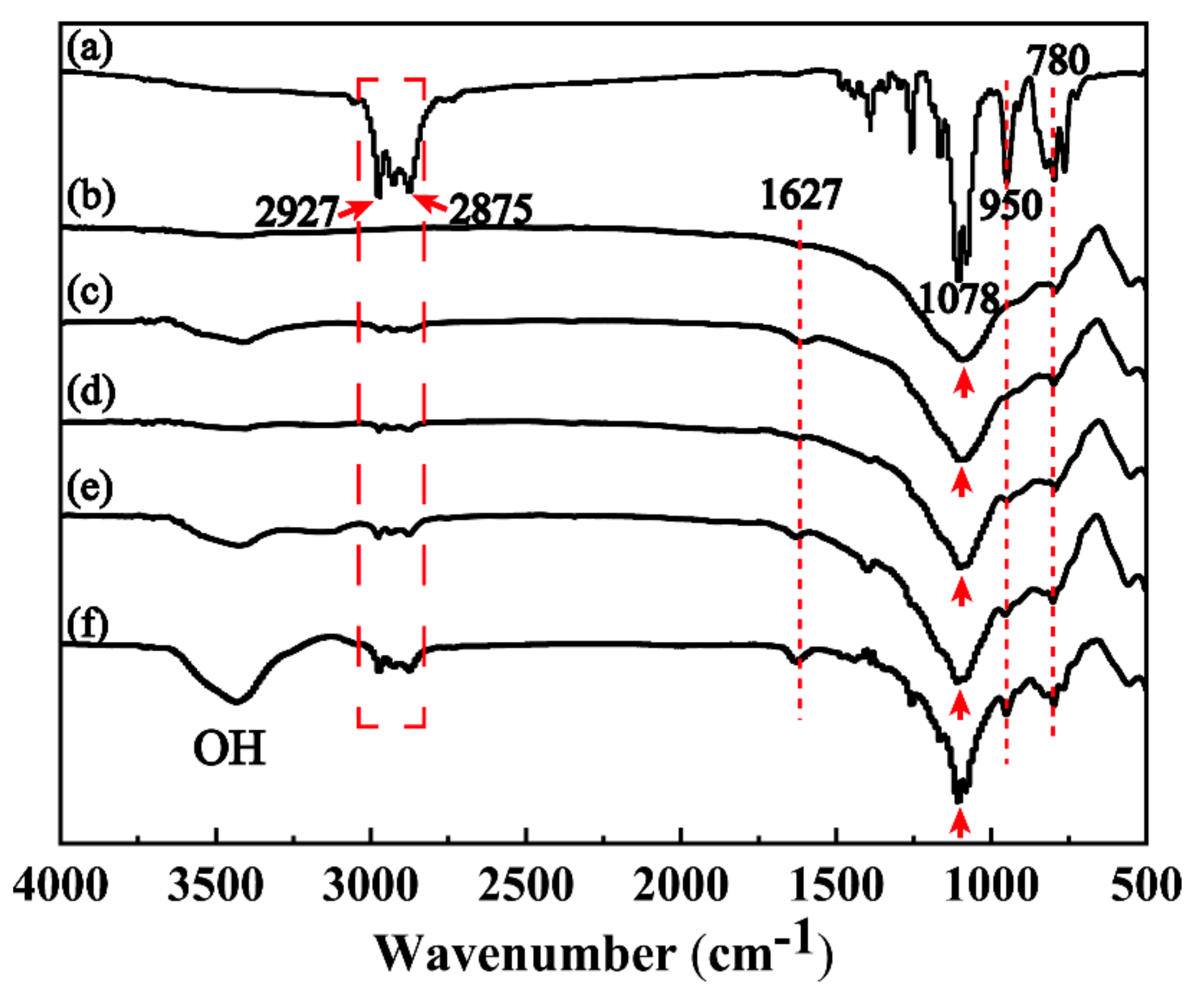
3.2. Modification and Optimization of MFA
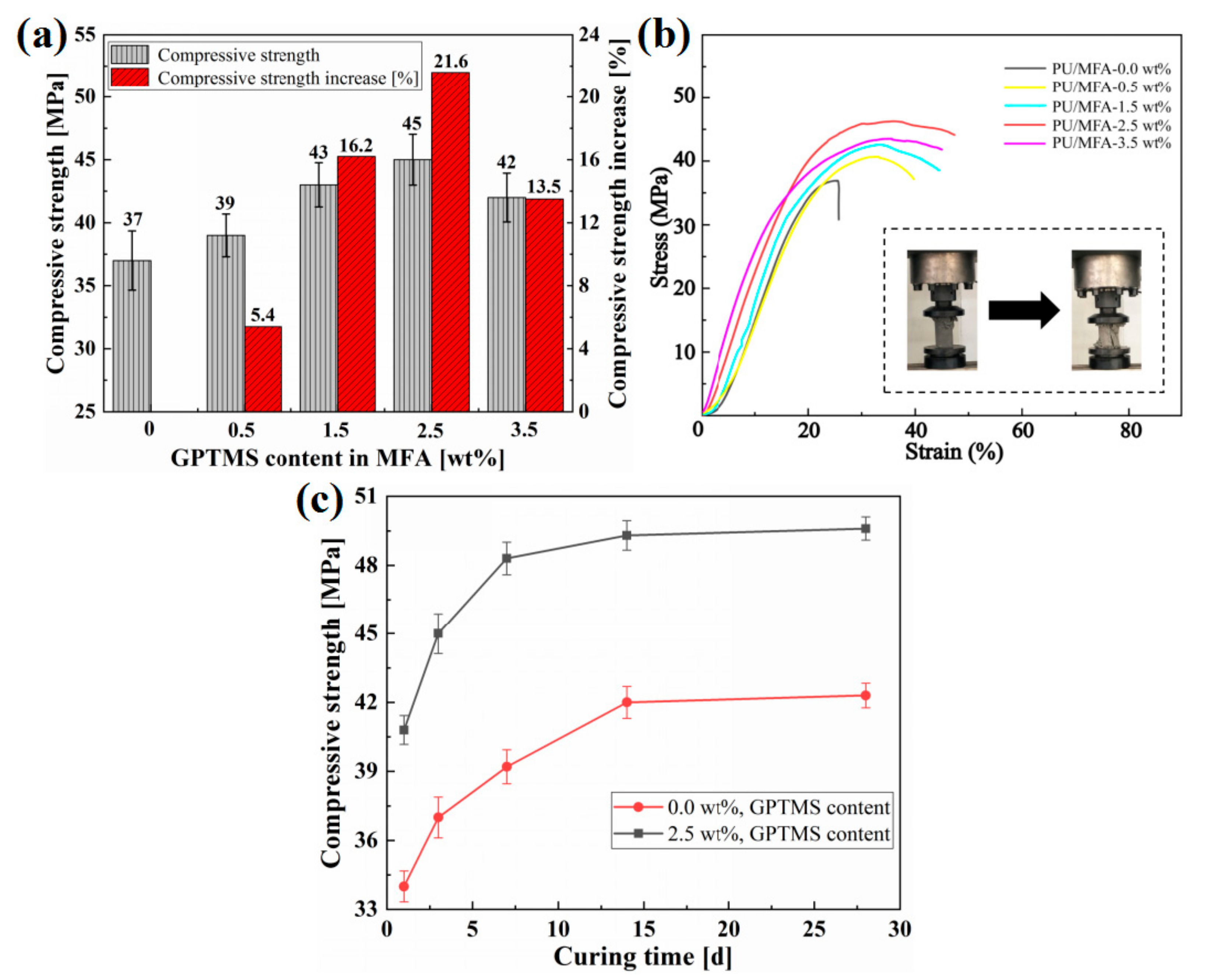
3.3. Compressive Strength of PU/MFA Composites
3.4. Fracture Toughness and Flexural Strength
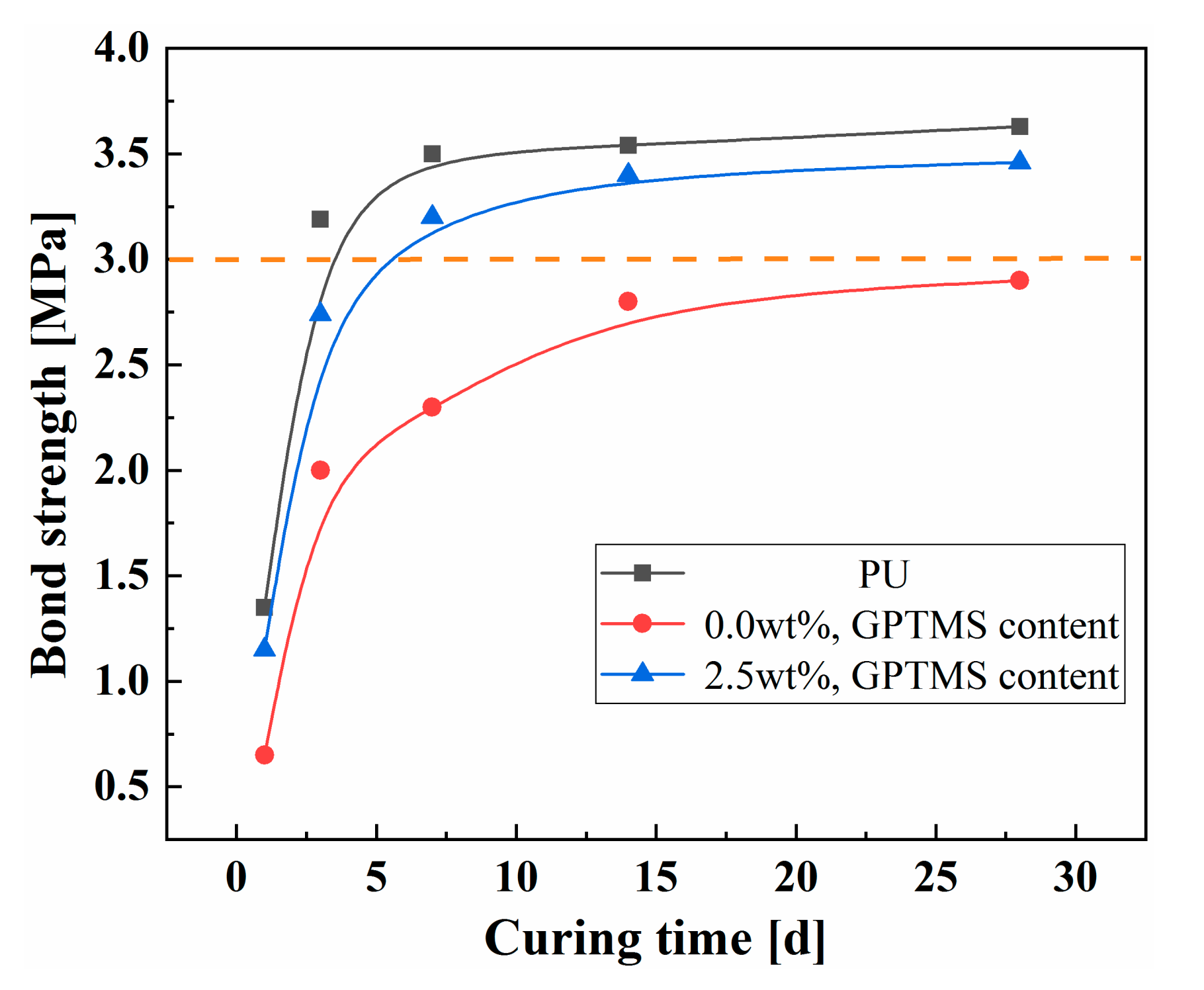
3.5. Bond Property Analysis
3.6. Fracture Surface Morphology of PU/MFA Materials
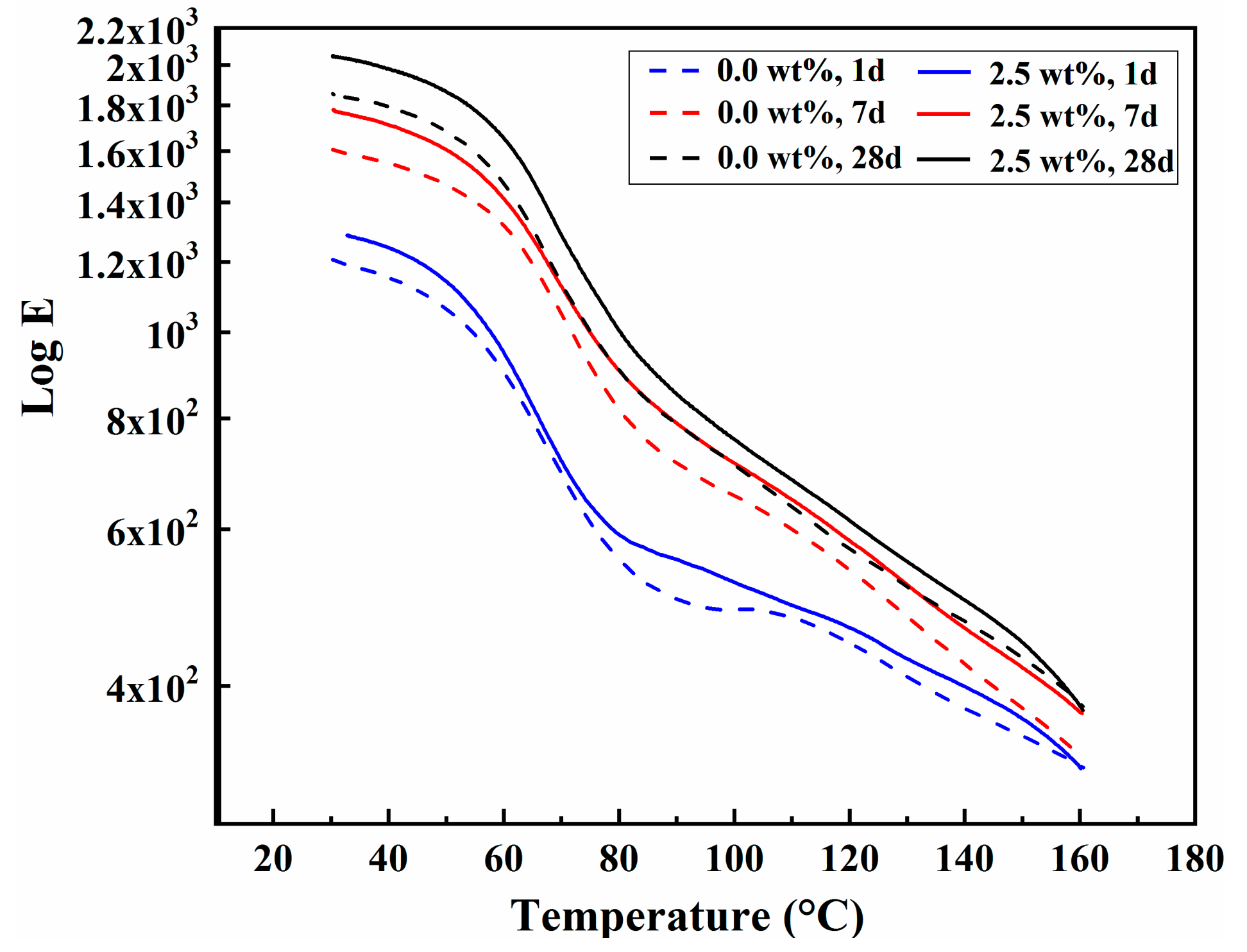
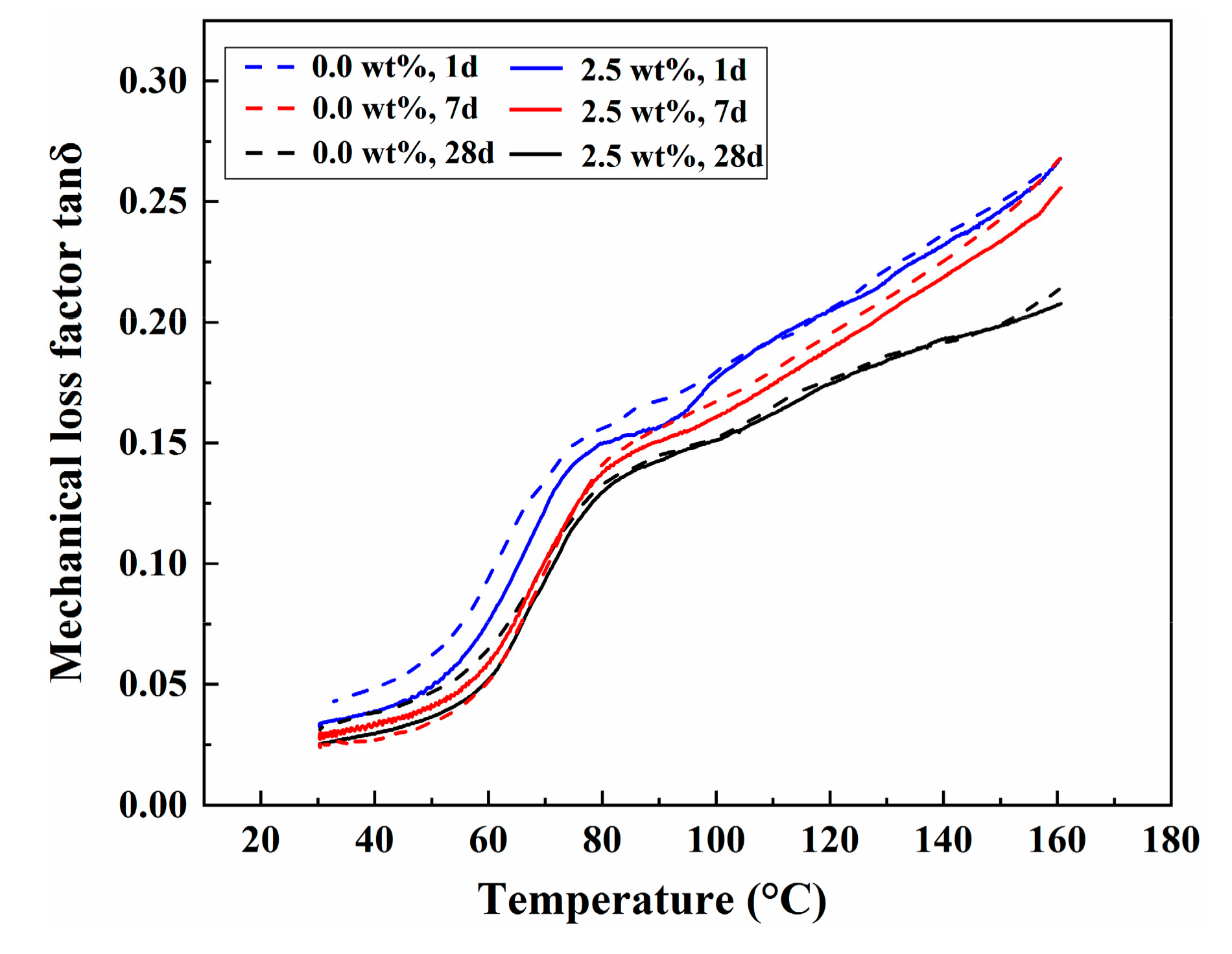
3.7. Dynamic Mechanical Analysis
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Cheng, C.; Chen, X.; Zhang, S. Multi-peak deformation behavior of jointed rock mass under uniaxial compression: Insight from particle flow modeling. Eng. Geol. 2016, 213, 25–45. [Google Scholar] [CrossRef]
- Zhang, C.; Fu, J.; Yang, J.; Ou, X.; Ye, X.; Zhang, Y. Formulation and performance of grouting materials for underwater shield tunnel construction in karst ground. Constr. Build. Mater. 2018, 187, 327–338. [Google Scholar] [CrossRef]
- Yang, Z.; Zhang, X.; Liu, X.; Guan, X.; Zhang, C.; Niu, Y. Flexible and stretchable polyurethane/waterglass grouting material. Constr. Build. Mater. 2017, 138, 240–246. [Google Scholar] [CrossRef]
- He, Z.; Li, Q.; Wang, J.; Yin, N.; Jiang, S.; Kang, M. Effect of silane treatment on the mechanical properties of polyurethane/water glass grouting materials. Constr. Build. Mater. 2016, 116, 110–120. [Google Scholar] [CrossRef]
- Besse, V.; Pluart, L.L.; Cook, W.D.; Pham, T.N.; Madec, P.J. Synthesis and polymerization kinetics of acrylamide phosphonic acids and esters as new dentine adhesives. J. Polym. Sci. Part A Polym. Chem. 2012, 51, 149–157. [Google Scholar] [CrossRef]
- Castella, N.; Grishchuk, S.; Karger-Kocsis, J.; Unik, M. Hybrid resins from polyisocyanate, epoxy resin and water glass: Chemistry, structure and properties. J. Mater. Sci. 2010, 45, 1734–1743. [Google Scholar] [CrossRef]
- Duan, H.; Jiang, Z.; Zhu, S.; Pu, Y.; Qiang, S. New composite grouting materials: Modified urea-formaldehyde resin with cement. Int. J. Min. Sci. Technol. 2012, 22, 195–200. [Google Scholar] [CrossRef]
- Heinzmann, C.; Salz, U.; Moszner, N.; Fiore, G.L.; Weder, C. Supramolecular cross-links in poly(alkyl methacrylate) copolymers and their impact on the mechanical and reversible adhesive properties. ACS Appl. Mater. Interfaces 2015, 7, 13395–13404. [Google Scholar] [CrossRef] [PubMed]
- Gutiérrez-González, S.; Gadea, J.; Rodríguez, A.; Junco, C.; Calderón, V. Lightweight plaster materials with enhanced thermal properties made with polyurethane foam wastes. Constr. Build. Mater. 2012, 28, 653–658. [Google Scholar] [CrossRef]
- Zhao, H. Development of water-soluble polyurethane grout. New Chem. Mater. 2005, 33, 8–9. [Google Scholar]
- Anagnostopoulos, C.A. Effect of different superplasticisers on the physical and mechanical properties of cement grouts. Constr. Build. Mater. 2014, 50, 162–168. [Google Scholar] [CrossRef]
- Tchakouté, H.K.; Rüscher, C.H.; Kong, S.; Kamseu, E.; Leonelli, C. Geopolymer binders from metakaolin using sodium waterglass from waste glass and rice husk ash as alternative activators: A comparative study. Constr. Build. Mater. 2016, 114, 276–289. [Google Scholar] [CrossRef]
- Wu, G.; Gu, J.; Zhao, X. Preparation and dynamic mechanical properties of polyurethane-modified epoxy composites filled with functionalized fly ash particulates. J. Appl. Polym. Sci. 2010, 105, 1118–1126. [Google Scholar] [CrossRef]
- Jung, S.H.; Saraswathy, V.; Karthick, S.; Kathirvel, P.; Kwon, S.J. Microstructure characteristics of fly ash concrete with rice husk ash and lime stone powder. Int. J. Concrete Struct. Mater. 2018, 12, 17. [Google Scholar] [CrossRef]
- Liu, G.W.; Li, S.P.; Gao, F.; Sun, W.Z.; Xiao, Z.J.; Yang, C.F. A foundational study on static mechanical characteristics of the super light weight and high strength material using fly-ash. J. Soc. Mater. Sci. 2018, 55, 738–741. [Google Scholar] [CrossRef]
- Rahman, M.M.; Han, D.K. Waterborne polyurethane/oil fly ash composite: A new environmentally friendly coating material. J. Adhes. Sci. Technol. 2015, 29, 2709–2718. [Google Scholar] [CrossRef]
- Hanemann, T.; Szabó, D.V. Polymer-Nanoparticle Composites: From Synthesis to Modern Applications. Materials 2010, 3, 3468–3517. [Google Scholar] [CrossRef]
- Zhao, J.; Milanova, M.; Warmoeskerken, M.M.C.G.; Dutschk, V. Surface modification of TiO2 nanoparticles with silane coupling agents. Colloids Surf. A Physicochem. Eng. Asp. 2012, 413, 273–279. [Google Scholar] [CrossRef]
- Dolatzadeh, F.; Moradian, S.; Jalili, M.M. Influence of various surface treated silica nanoparticles on the electrochemical properties of SiO/polyurethane nanocoatings. Corros. Sci. 2011, 53, 4248–4257. [Google Scholar] [CrossRef]
- Kango, S.; Kalia, S.; Celli, A.; Njuguna, J.; Habibi, Y.; Kumar, R. Surface modification of inorganic nanoparticles for development of organic–inorganic nanocomposites—A review. Prog. Polym. Sci. 2013, 38, 1232–1361. [Google Scholar] [CrossRef]
- Xie, J.; Wu, S.; Pang, L.; Lin, J.; Zhu, Z. Influence of surface treated fly ash with coupling agent on asphalt mixture moisture damage. Constr. Build. Mater. 2012, 30, 340–346. [Google Scholar] [CrossRef]
- Parvaiz, M.R.; Mohanty, S.; Nayak, S.K.; Mahanwar, P.A. Effect of surface modification of fly ash on the mechanical, thermal, electrical and morphological properties of polyetheretherketone composites. Mater. Sci. Eng. A. 2011, 528, 4277–4286. [Google Scholar] [CrossRef]
- Ishikawa, T.; Ejaz, M.; Tsujii, Y.; Shibata, H.; Matsumoto, M. Phase-separated structures of mixed LB films of silane-coupling agents with polymerization initiating groups and amphiphilic carboxylic acids. Colloids Surf. A Physicochem. Eng. Asp. 2008, 321, 76–81. [Google Scholar] [CrossRef]
- Dun, Y.; Zuo, Y. Preparation and characterization of a GPTMS/graphene coating on AA-2024 alloy. Appl. Surf. Sci. 2017, 416, 492–502. [Google Scholar] [CrossRef]
- Rao, M.G.S.; Sánchez-Martinez, A.; Gutiérrez-Heredia, G.; Quevedo-López, M.A.; Ramírez-Bon, R. Sol-gel derived low temperature HfO2-GPTMS hybrid gate dielectric for a-IGZO thin-film transistors (TFTs). Ceram. Int. 2018, 44, 16428–16434. [Google Scholar] [CrossRef]
- Guido, E.; Colleoni, C.; Clerck, K.D.; Plutino, M.R.; Rosace, G. Influence of catalyst in the synthesis of a cellulose-based sensor: Kinetic study of 3-glycidoxypropyltrimethoxysilane epoxy ring opening by Lewis acid. Sens. Actuators B Chem. 2014, 203, 213–222. [Google Scholar] [CrossRef]
- Innocenzi, P.; Brusatin, G. Competitive polymerization between organic and inorganic networks in hybrid materials. Chem. Mater. 2000, 12, 3726–3732. [Google Scholar] [CrossRef]
- Baltazar, L.G.; Henriques, F.M.A.; Jorne, F.; Cidade, M.T. Combined effect of superplasticizer, silica fume and temperature in the performance of natural hydraulic lime grouts. Constr. Build. Mater. 2014, 50, 584–597. [Google Scholar] [CrossRef]
- Chaowasakoo, T.; Sombatsompop, N. Mechanical and morphological properties of fly ash/epoxy composites using conventional thermal and microwave curing methods. Compos. Sci. Technol. 2007, 67, 2282–2291. [Google Scholar] [CrossRef]
- Culler, S.R.; Ishida, H.; Koenig, J.L. FT-IR characterization of the reaction at the silane/matrix resin interphase of composite materials. J. Colloid Interface Sci. 1986, 109, 1–10. [Google Scholar] [CrossRef]
- Idris, A.; Man, Z.; Maulud, A.S.; Mannan, H.A.; Shafie, A. Effect of silane coupling agents on properties and performance of polycarbonate/silica MMMs. PolymTest. 2019, 73, 159–170. [Google Scholar] [CrossRef]
- Yang, Y.Y.; Wang, J.Q.; Dou, H.J. Mechanical properties of anti-seepage grouting materials for heavy metal contaminated soil. Trans. Nonferrous Metals Soc. China. 2014, 24, 3316–3323. [Google Scholar] [CrossRef]
- Yuan, Q.; Caillard, J.; Rong, L. Fracture toughness of soft materials with rate-independent hysteresis. J. Mech. Phys. Solids. 2018, 118, 341–364. [Google Scholar]
- Bagheri, R.; Pearson, R.A. Role of particle cavitation in rubber-toughened epoxies: II. Inter-particle distance. Polymer 2000, 41, 269–276. [Google Scholar] [CrossRef]
- Sun, J.; Fu, Q.G.; Huo, C.X.; Li, T. Fracture toughness of thermally sprayed MoSi2 composite with different melting indices. Compos. Part B Eng. 2018, 150, 242–247. [Google Scholar] [CrossRef]
- Wang, J.; Xue, Z.; Li, Y.; Li, G.; Wang, Y.; Zhong, W.H. Synergistically effects of copolymer and core-shell particles for toughening epoxy. Polymer. 2018, 140, 39–46. [Google Scholar] [CrossRef]
- Yousefi, E.; Ghadimi, M.R.; Amirpoor, S.; Dolati, A. Preparation of new superhydrophobic and highly oleophobic polyurethane coating with enhanced mechanical durability. Appl. Surf. Sci. 2018, 454, 201–209. [Google Scholar] [CrossRef]
- Haghdadeh, P.; Ghaffari, M.; Ramezanzadeh, B.; Bahlakeh, G.; Saeb, M.R. The role of functionalized graphene oxide on the mechanical and anti-corrosion properties of polyurethane coating. J. Taiwan Inst. Chem. Eng. 2018, 86, 199–212. [Google Scholar] [CrossRef]
- Cai, X.; Zhang, H.; Zhang, J.; Chen, X.; Yang, J.; Hong, J. Investigation on reinforcing mechanisms of semi-flexible pavement material through micromechanical model. Constr. Build. Mater. 2019, 198, 732–741. [Google Scholar] [CrossRef]
- Koutsoumpis, S.; Ozimek, J.; Raftopoulos, K.N.; Hebda, E.; Klonos, P.; Papadakis, C.M. Polyurethanes with POSS pendent on flexible hard segments: Morphology and glass transition. Polymer 2018, 147, 225–236. [Google Scholar] [CrossRef]
- Lee, D.-J.; Kim, M.-K.; Walsh, J.; Jang, H.-K.; Kim, H.-I.; Oh, E.-Y. Experimental characterization of temperature dependent dynamic properties of glass fiber reinforced polyurethane foams. Polym. Test. 2019, 74, 30–38. [Google Scholar] [CrossRef]
- Hu, Z.-X.; Hu, X.M.; Cheng, W.-M.; Zhao, Y.-Y.; Wu, M.-Y. Performance optimization of one-component polyurethane healing agent for self-healing concrete. Constr. Build. Mater. 2018, 179, 151–159. [Google Scholar] [CrossRef]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Composite | Ingredient | Amount (g) | Manufacturer | Function | Properties |
|---|---|---|---|---|---|
| Component A | The polyether polyol (GR4110B) | 90 ± 0.1 | Shanghai Gaoqiao Petrochemica Co., Ltd. | Reactants | Molecular weight (g/mol): 1500 |
| Viscosity (mPa.s): 6000–9000 | |||||
| Hydroxyl value (mg KOH/g): 450 | |||||
| Moisture (%): ≤0.2 | |||||
| The polyether polyol (TMN450) | 10 ± 0.1 | Jiangsu Haian Petrochemical Plant | Reactants | Molecular weight (g/mol): 1300 | |
| Viscosity (mPa·s): 483 | |||||
| Hydroxyl value (mg KOH/g): 460 | |||||
| Moisture (%): ≤0.1 | |||||
| Stannous octanate | 0.3 ± 0.02 | Shandong Baidu Chemical Co., Ltd. | Catalyzer | Molecular weight (g/mol): 327.56 | |
| Proportion (20 °C): 1.27~1.31 | |||||
| MFA | 140.2 ± 0.1 | Huaneng Jinling Power Plant | Aggregate | Moisture (%): ≤0.2 | |
| Particle size (nm): ≤10 | |||||
| Chloride ion (%): 0.01 | |||||
| GPTMS | 0.7~4.9 | Nanjing Nengde Chemical Industry Co., Ltd. | Silane coupling agent | Moisture (%): ≤0.5 | |
| Tris (2-chloro-1-methylethyl) phosphate (TCPP) | 1 ± 0.02 | Terry New Materials Co., Ltd. | Flame retardant | ||
| Component B | Polymethylene polyphenylene isocyanate (PAPI) | 100 ± 0.1 | Polyurethane Co., Ltd. | Reactants | Molecular weight (g/mol): 1600 |
| NCO content (%): 30 | |||||
| Viscosity (mPa·s): 150–250 | |||||
| Density (g/mol): 1.25 | |||||
| Acidity (%): ≤0.05 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Qin, C.; Lu, W.; He, Z.; Qi, G.; Li, J.; Hu, X. Effect of Silane Treatment on Mechanical Properties of Polyurethane/Mesoscopic Fly Ash Composites. Polymers 2019, 11, 741. https://doi.org/10.3390/polym11040741
Qin C, Lu W, He Z, Qi G, Li J, Hu X. Effect of Silane Treatment on Mechanical Properties of Polyurethane/Mesoscopic Fly Ash Composites. Polymers. 2019; 11(4):741. https://doi.org/10.3390/polym11040741
Chicago/Turabian StyleQin, Chuanrui, Wei Lu, Zhenglong He, Guansheng Qi, Jinliang Li, and Xiangming Hu. 2019. "Effect of Silane Treatment on Mechanical Properties of Polyurethane/Mesoscopic Fly Ash Composites" Polymers 11, no. 4: 741. https://doi.org/10.3390/polym11040741