Advances on Non-Genetic Cell Membrane Engineering for Biomedical Applications


Abstract
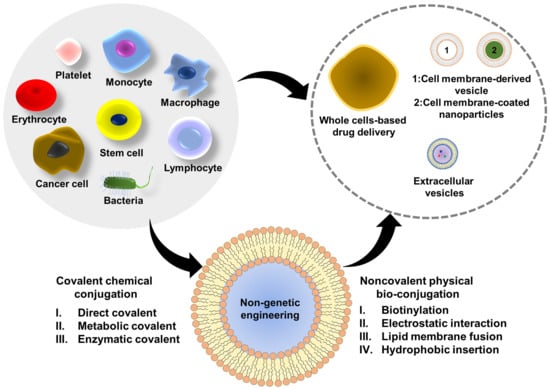
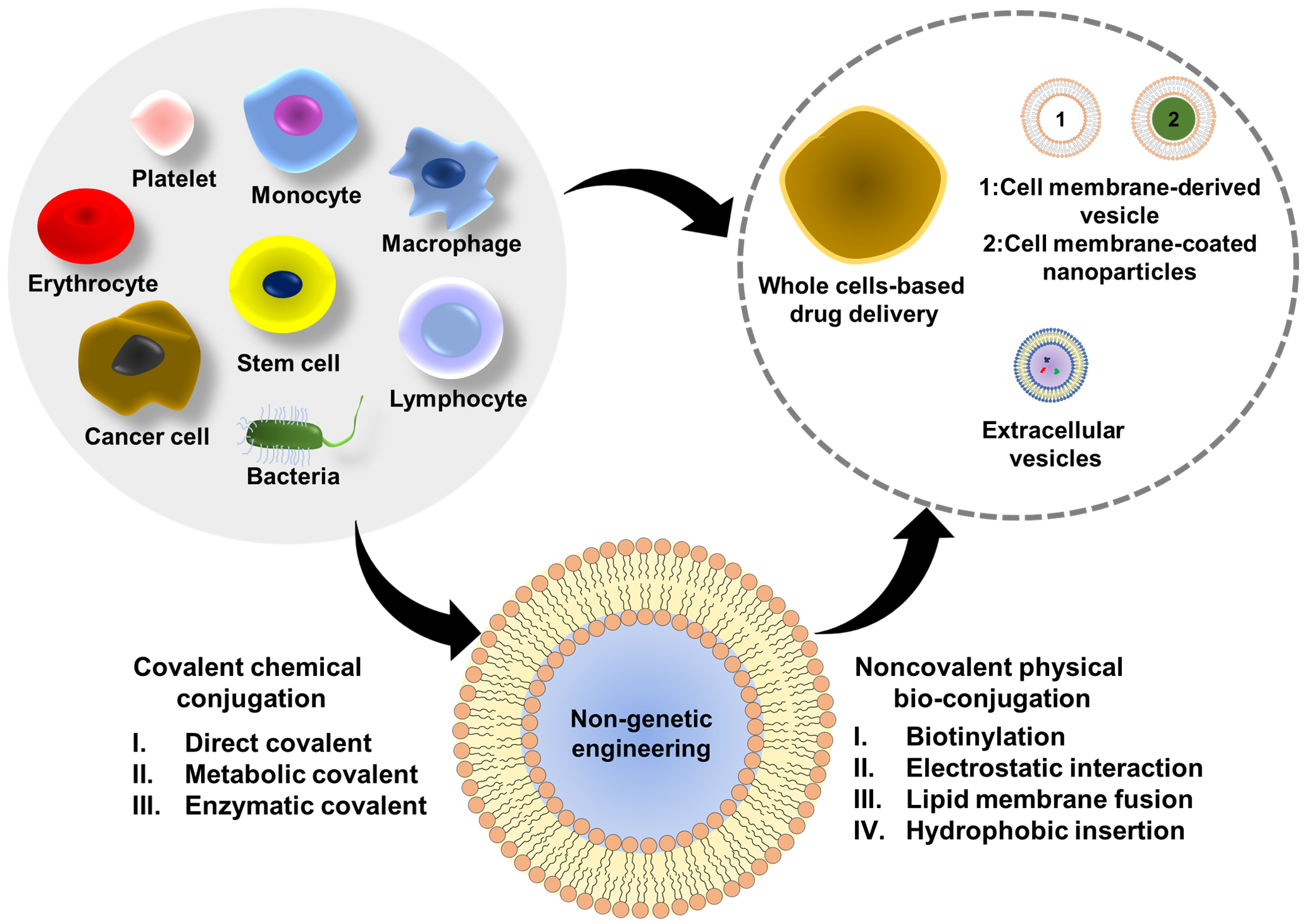
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Non-Genetic Surface Engineering Strategies
2.1. Covalent Chemical Conjugations
2.2. Noncovalent Physical Bioconjugations
3. Application of Cell Surface Engineering in Different Cells or Cell-Based Therapeutics
3.1. Erythrocytes

3.2. Platelets
3.3. Cancer Cells
3.4. Monocytes/Macrophages
3.5. Lymphocytes
3.6. Stem Cells
3.7. Bacteria
3.8. Extracellular Vesicles

4. Perspective and Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Pierigè, F.; Serafini, S.; Rossi, L.; Magnani, M. Cell-based drug delivery. Adv. Drug Deliv. Rev. 2008, 60, 286–295. [Google Scholar] [CrossRef] [PubMed]
- Fliervoet, L.A.; Mastrobattista, E. Drug delivery with living cells. Adv. Drug Deliv. Rev. 2016, 106, 63–72. [Google Scholar] [CrossRef] [PubMed]
- Fischbach, M.A.; Bluestone, J.A.; Lim, W.A. Cell-based therapeutics: The next pillar of medicine. Sci. Transl. Med. 2013, 5, 179ps7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Villa, C.H.; Cines, D.B.; Siegel, D.L.; Muzykantov, V. Erythrocytes as carriers for drug delivery in blood transfusion and beyond. Transfus. Med. Rev. 2017, 31, 26–35. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Biagiotti, S.; Paoletti, M.F.; Fraternale, A.; Rossi, L.; Magnani, M. Drug delivery by red blood cells. IUBMB Life 2011, 63, 621–631. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.R.; Zhang, D.; Oswald, B.E.; Carrim, N.; Wang, X.; Hou, Y.; Zhang, Q.; Lavalle, C.; McKeown, T.; Marshall, A.H. Platelets are versatile cells: New discoveries in hemostasis, thrombosis, immune responses, tumor metastasis and beyond. Crit. Rev. Clin. Lab. Sci. 2016, 53, 409–430. [Google Scholar] [CrossRef] [PubMed]
- Franco, A.T.; Corken, A.; Ware, J. Platelets at the interface of thrombosis, inflammation, and cancer. Blood 2015, 126, 582–588. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fang, R.H.; Kroll, A.V.; Gao, W.; Zhang, L. Cell membrane coating nanotechnology. Adv. Mater. 2018, 30, e1706759. [Google Scholar] [CrossRef]
- Ahmed, K.K.; Geary, S.M.; Salem, A.K. Surface engineering tumor cells with adjuvant-loaded particles for use as cancer vaccines. J. Control. Release 2017, 248, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Stephan, M.T.; Irvine, D.J. Enhancing cell therapies from the outside in: Cell surface engineering using synthetic nanomaterials. Nano Today 2011, 6, 309–325. [Google Scholar] [CrossRef] [Green Version]
- Morgan, R.A.; Dudley, M.E.; Wunderlich, J.R.; Hughes, M.S.; Yang, J.C.; Sherry, R.M.; Royal, R.E.; Topalian, S.L.; Kammula, U.S.; Restifo, N.P. Cancer regression in patients after transfer of genetically engineered lymphocytes. Science 2006, 314, 126–129. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tran, C.; Damaser, M.S. Stem cells as drug delivery methods: Application of stem cell secretome for regeneration. Adv. Drug Deliv. Rev. 2015, 82, 1–11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ankrum, J.; Karp, J.M. Mesenchymal stem cell therapy: Two steps forward, one step back. Trends Mol. Med. 2010, 16, 203–209. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karp, J.M.; Teo, G.S.L. Mesenchymal stem cell homing: The devil is in the details. Cell Stem Cell 2009, 4, 206–216. [Google Scholar] [CrossRef] [Green Version]
- Sarkar, D.; Vemula, P.K.; Zhao, W.; Gupta, A.; Karnik, R.; Karp, J.M. Engineered mesenchymal stem cells with self-assembled vesicles for systemic cell targeting. Biomaterials 2010, 31, 5266–5274. [Google Scholar] [CrossRef] [Green Version]
- Suh, S.; Jo, A.; Traore, M.A.; Zhan, Y.; Coutermarsh-Ott, S.L.; Ringel-Scaia, V.M.; Allen, I.C.; Davis, R.M.; Behkam, B. Nanoscale Bacteria-Enabled Autonomous Drug Delivery System (NanoBEADS) Enhances Intratumoral Transport of Nanomedicine. Adv. Sci. 2019, 6, 1801309. [Google Scholar] [CrossRef] [Green Version]
- Akin, D.; Sturgis, J.; Ragheb, K.; Sherman, D.; Burkholder, K.; Robinson, J.P.; Bhunia, A.K.; Mohammed, S.; Bashir, R. Bacteria-mediated delivery of nanoparticles and cargo into cells. Nat. Nanotechnol. 2007, 2, 441–449. [Google Scholar] [CrossRef] [Green Version]
- Parodi, A.; Molinaro, R.; Sushnitha, M.; Evangelopoulos, M.; Martinez, J.O.; Arrighetti, N.; Corbo, C.; Tasciotti, E. Bio-inspired engineering of cell-and virus-like nanoparticles for drug delivery. Biomaterials 2017, 147, 155–168. [Google Scholar] [CrossRef]
- Wang, Q.; Cheng, H.; Peng, H.; Zhou, H.; Li, P.Y.; Langer, R. Non-genetic engineering of cells for drug delivery and cell-based therapy. Adv. Drug Deliv. Rev. 2015, 91, 125–140. [Google Scholar] [CrossRef]
- Zhai, Y.; Su, J.; Ran, W.; Zhang, P.; Yin, Q.; Zhang, Z.; Yu, H.; Li, Y. Preparation and application of cell membrane-camouflaged nanoparticles for cancer therapy. Theranostics 2017, 7, 2575–2592. [Google Scholar] [CrossRef]
- Armstrong, J.P.; Holme, M.N.; Stevens, M.M. Re-engineering extracellular vesicles as smart nanoscale therapeutics. ACS Nano 2017, 11, 69–83. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Richardson, J.J.; Ejima, H. Surface Engineering of Extracellular Vesicles through Chemical and Biological Strategies. Chem. Mater. 2019, 31, 2191–2201. [Google Scholar] [CrossRef]
- Thanuja, M.; Anupama, C.; Ranganath, S.H. Bioengineered cellular and cell membrane-derived vehicles for actively targeted drug delivery: So near and yet so far. Adv. Drug Deliv. Rev. 2018, 132, 57–80. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Hu, Q.; Gu, Z. Leveraging engineering of cells for drug delivery. Acc. Chem. Res. 2018, 51, 668–677. [Google Scholar] [CrossRef]
- Teramura, Y.; Iwata, H. Cell surface modification with polymers for biomedical studies. Soft Matter 2010, 6, 1081–1091. [Google Scholar] [CrossRef] [Green Version]
- Park, J.; Andrade, B.; Seo, Y.; Kim, M.-J.; Zimmerman, S.C.; Kong, H. Engineering the surface of therapeutic “living” cells. Chem. Rev. 2018, 118, 1664–1690. [Google Scholar] [CrossRef]
- Hayashi, Y.; Tsuji, S.; Tsujii, M.; Nishida, T.; Ishii, S.; Iijima, H.; Nakamura, T.; Eguchi, H.; Miyoshi, E.; Hayashi, N. Topical implantation of mesenchymal stem cells has beneficial effects on healing of experimental colitis in rats. J. Pharmacol. Exp. Ther. 2008, 326, 523–531. [Google Scholar] [CrossRef] [Green Version]
- Fox, W.M.; Sarkar, D. Cell Surface Engineering by Chemical Reaction and Remodeling. In Micro- and Nanoengineering of the Cell Surface, 1st ed.; Karp, J.M., Zhao, W., Eds.; William Andrew: Oxford, UK, 2014; pp. 27–41. [Google Scholar]
- Custódio, C.A.; Mano, J.F. Cell surface engineering to control cellular interactions. ChemNanoMat 2016, 2, 376–384. [Google Scholar] [CrossRef]
- June, C.H.; O’Connor, R.S.; Kawalekar, O.U.; Ghassemi, S.; Milone, M.C. CAR T cell immunotherapy for human cancer. Science 2018, 359, 1361–1365. [Google Scholar] [CrossRef] [Green Version]
- Abbina, S.; Siren, E.M.; Moon, H.; Kizhakkedathu, J.N. Surface engineering for cell-based therapies: Techniques for manipulating mammalian cell surfaces. ACS Biomater. Sci. Eng. 2017, 4, 3658–3677. [Google Scholar] [CrossRef]
- Kim, J.C.; Tae, G. Recent advances in cell surface engineering focused on cell therapy. Bull. Korean Chem. Soc. 2015, 36, 59–65. [Google Scholar] [CrossRef]
- Armstrong, J.P.; Perriman, A.W. Strategies for cell membrane functionalization. Exp. Biol. Med. 2016, 241, 1098–1106. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Csizmar, C.M.; Petersburg, J.R.; Wagner, C.R. Programming cell-cell interactions through non-genetic membrane engineering. Cell Chem. Biol. 2018, 25, 931–940. [Google Scholar] [CrossRef] [PubMed]
- Lee, D.Y.; Cha, B.-H.; Jung, M.; Kim, A.S.; Bull, D.A.; Won, Y.-W. Cell surface engineering and application in cell delivery to heart diseases. J. Biol. Eng. 2018, 12, 28. [Google Scholar] [CrossRef] [PubMed]
- O’Brien, P.J.; Elahipanah, S.; Rogozhnikov, D.; Yousaf, M.N. Bio-orthogonal mediated nucleic acid transfection of cells via cell surface engineering. ACS Cent. Sci. 2017, 3, 489–500. [Google Scholar] [CrossRef] [PubMed]
- Zhang, P.; Bookstaver, M.L.; Jewell, C.M. Engineering cell surfaces with polyelectrolyte materials for translational applications. Polymers 2017, 9, 40. [Google Scholar] [CrossRef] [Green Version]
- Han, X.; Wang, C.; Liu, Z. Red blood cells as smart delivery systems. Bioconjug. Chem. 2018, 29, 852–860. [Google Scholar] [CrossRef]
- Scott, M.D.; Murad, K.L.; Koumpouras, F.; Talbot, M.; Eaton, J.W. Chemical camouflage of antigenic determinants: Stealth erythrocytes. Proc. Natl. Acad. Sci. USA 1997, 94, 7566–7571. [Google Scholar] [CrossRef] [Green Version]
- Chiarantini, L.; Droleskey, R.; Magnani, M.; DeLoach, J. In vitro targeting of erythrocytes to cytotoxic T-cells by coupling of Thy-1.2 monoclonal antibody. Biotechnol. Appl. Biochem 1992, 15, 171–184. [Google Scholar]
- Kirch, H. Enhanced biological activity of human recombinant interleukin 2 coupled to mouse red blood cells as evaluated using the mouse Meth A sarcoma model. Biotechnol. Appl. Biochem. 1996, 23, 29–36. [Google Scholar]
- Spitzer, D.; Unsinger, J.; Bessler, M.; Atkinson, J.P. ScFv-mediated in vivo targeting of DAF to erythrocytes inhibits lysis by complement. Mol. Immunol. 2004, 40, 911–919. [Google Scholar] [CrossRef] [PubMed]
- Stein, S.C.; Ganguly, K.; Belfield, C.M.; Xu, X.; Swanson, E.W.; Chen, X.-H.; Browne, K.D.; Johnson, V.E.; Smith, D.H.; LeBold, D.G. Erythrocyte-bound tissue plasminogen activator is neuroprotective in experimental traumatic brain injury. J. Neurotrauma 2009, 26, 1585–1592. [Google Scholar] [CrossRef] [PubMed]
- Rossi, N.A.; Constantinescu, I.; Kainthan, R.K.; Brooks, D.E.; Scott, M.D.; Kizhakkedathu, J.N. Red blood cell membrane grafting of multi-functional hyperbranched polyglycerols. Biomaterials 2010, 31, 4167–4178. [Google Scholar] [CrossRef] [PubMed]
- Chessa, L.; Leuzzi, V.; Plebani, A.; Soresina, A.; Micheli, R.; D’Agnano, D.; Venturi, T.; Molinaro, A.; Fazzi, E.; Marini, M. Intra-erythrocyte infusion of dexamethasone reduces neurological symptoms in ataxia teleangiectasia patients: Results of a phase 2 trial. Orphanet J. Rare Dis. 2014, 9, 5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bax, B.E.; Bain, M.D.; Scarpelli, M.; Filosto, M.; Tonin, P.; Moran, N. Clinical and biochemical improvements in a patient with MNGIE following enzyme replacement. Neurology 2013, 81, 1269–1271. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rossi, L.; Pierigè, F.; Carducci, C.; Gabucci, C.; Pascucci, T.; Canonico, B.; Bell, S.M.; Fitzpatrick, P.A.; Leuzzi, V.; Magnani, M. Erythrocyte-mediated delivery of phenylalanine ammonia lyase for the treatment of phenylketonuria in BTBR-Pahenu2 mice. J. Control. Release 2014, 194, 37–44. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anselmo, A.C.; Gupta, V.; Zern, B.J.; Pan, D.; Zakrewsky, M.; Muzykantov, V.; Mitragotri, S. Delivering nanoparticles to lungs while avoiding liver and spleen through adsorption on red blood cells. ACS Nano 2013, 7, 11129–11137. [Google Scholar] [CrossRef] [Green Version]
- Chambers, E.; Mitragotri, S. Prolonged circulation of large polymeric nanoparticles by non-covalent adsorption on erythrocytes. J. Control. Release 2004, 100, 111–119. [Google Scholar] [CrossRef]
- Murciano, J.-C.; Higazi, A.A.-R.; Cines, D.B.; Muzykantov, V.R. Soluble urokinase receptor conjugated to carrier red blood cells binds latent pro-urokinase and alters its functional profile. J. Control. Release 2009, 139, 190–196. [Google Scholar] [CrossRef] [Green Version]
- Sun, X.; Han, X.; Xu, L.; Gao, M.; Xu, J.; Yang, R.; Liu, Z. Surface-Engineering of Red Blood Cells as Artificial Antigen Presenting Cells Promising for Cancer Immunotherapy. Small 2017, 13, 1701864. [Google Scholar] [CrossRef]
- Rao, L.; Meng, Q.-F.; Bu, L.-L.; Cai, B.; Huang, Q.; Sun, Z.-J.; Zhang, W.-F.; Li, A.; Guo, S.-S.; Liu, W. Erythrocyte membrane-coated upconversion nanoparticles with minimal protein adsorption for enhanced tumor imaging. ACS Appl. Mater. Interfaces 2017, 9, 2159–2168. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Ye, Y.; Sun, W.; Yu, J.; Wang, J.; Lawrence, D.S.; Buse, J.B.; Gu, Z. Red blood cells for glucose-responsive insulin delivery. Adv. Mater. 2017, 29, 1606617. [Google Scholar] [CrossRef] [PubMed]
- Murciano, J.-C.; Medinilla, S.; Eslin, D.; Atochina, E.; Cines, D.B.; Muzykantov, V.R. Prophylactic fibrinolysis through selective dissolution of nascent clots by tPA-carrying erythrocytes. Nat. Biotechnol. 2003, 21, 891–896. [Google Scholar] [CrossRef] [PubMed]
- Zaitsev, S.; Danielyan, K.; Murciano, J.-C.; Ganguly, K.; Krasik, T.; Taylor, R.P.; Pincus, S.; Jones, S.; Cines, D.B.; Muzykantov, V.R. Human complement receptor type 1—Directed loading of tissue plasminogen activator on circulating erythrocytes for prophylactic fibrinolysis. Blood 2006, 108, 1895–1902. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, C.; Sun, X.; Cheng, L.; Yin, S.; Yang, G.; Li, Y.; Liu, Z. Multifunctional theranostic red blood cells for magnetic-field-enhanced in vivo combination therapy of cancer. Adv. Mater. 2014, 26, 4794–4802. [Google Scholar] [CrossRef]
- Fang, R.H.; Hu, C.-M.J.; Chen, K.N.; Luk, B.T.; Carpenter, C.W.; Gao, W.; Li, S.; Zhang, D.-E.; Lu, W.; Zhang, L. Lipid-insertion enables targeting functionalization of erythrocyte membrane-cloaked nanoparticles. Nanoscale 2013, 5, 8884–8888. [Google Scholar] [CrossRef] [Green Version]
- Guo, Y.; Wang, D.; Song, Q.; Wu, T.; Zhuang, X.; Bao, Y.; Kong, M.; Qi, Y.; Tan, S.; Zhang, Z. Erythrocyte membrane-enveloped polymeric nanoparticles as nanovaccine for induction of antitumor immunity against melanoma. ACS Nano 2015, 9, 6918–6933. [Google Scholar] [CrossRef]
- Korin, N.; Kanapathipillai, M.; Matthews, B.D.; Crescente, M.; Brill, A.; Mammoto, T.; Ghosh, K.; Jurek, S.; Bencherif, S.A.; Bhatta, D. Shear-activated nanotherapeutics for drug targeting to obstructed blood vessels. Science 2012, 337, 738–742. [Google Scholar] [CrossRef] [Green Version]
- Gay, L.J.; Felding-Habermann, B. Contribution of platelets to tumour metastasis. Nat. Rev. Cancer 2011, 11, 123–134. [Google Scholar] [CrossRef]
- Labelle, M.; Hynes, R.O. The initial hours of metastasis: The importance of cooperative host-tumor cell interactions during hematogenous dissemination. Cancer Discov. 2012, 2, 1091–1099. [Google Scholar] [CrossRef] [Green Version]
- Hu, Q.; Sun, W.; Qian, C.; Wang, C.; Bomba, H.N.; Gu, Z. Anticancer platelet-mimicking nanovehicles. Adv. Mater. 2015, 27, 7043–7050. [Google Scholar] [CrossRef]
- Hu, Q.; Qian, C.; Sun, W.; Wang, J.; Chen, Z.; Bomba, H.N.; Xin, H.; Shen, Q.; Gu, Z. Engineered nanoplatelets for enhanced treatment of multiple myeloma and thrombus. Adv. Mater. 2016, 28, 9573–9580. [Google Scholar] [CrossRef] [Green Version]
- Wang, C.; Sun, W.; Ye, Y.; Hu, Q.; Bomba, H.N.; Gu, Z. In situ activation of platelets with checkpoint inhibitors for post-surgical cancer immunotherapy. Nat. Biomed. Eng. 2017, 1, 0011. [Google Scholar] [CrossRef]
- Hu, Q.; Sun, W.; Wang, J.; Ruan, H.; Zhang, X.; Ye, Y.; Shen, S.; Wang, C.; Lu, W.; Cheng, K. Conjugation of haematopoietic stem cells and platelets decorated with anti-PD-1 antibodies augments anti-leukaemia efficacy. Nat. Biomed. Eng. 2018, 2, 831. [Google Scholar] [CrossRef]
- Shirota, H.; Klinman, D.M. CpG-conjugated apoptotic tumor cells elicit potent tumor-specific immunity. Cancer Immunol. Immunother. 2011, 60, 659–669. [Google Scholar] [CrossRef]
- Shi, P.; Ju, E.; Yan, Z.; Gao, N.; Wang, J.; Hou, J.; Zhang, Y.; Ren, J.; Qu, X. Spatiotemporal control of cell–cell reversible interactions using molecular engineering. Nat. Commun. 2016, 7, 13088. [Google Scholar] [CrossRef] [Green Version]
- Wang, H.; Wang, R.; Cai, K.; He, H.; Liu, Y.; Yen, J.; Wang, Z.; Xu, M.; Sun, Y.; Zhou, X. Selective in vivo metabolic cell-labeling-mediated cancer targeting. Nat. Chem. Biol. 2017, 13, 415–424. [Google Scholar] [CrossRef]
- Aizik, G.; Grad, E.; Golomb, G. Monocyte-mediated drug delivery systems for the treatment of cardiovascular diseases. Drug Deliv. Transl. Res. 2018, 8, 868–882. [Google Scholar] [CrossRef]
- Sabir, F.; Farooq, R.K.; Ahmed, N. Monocyte as an Emerging Tool for Targeted Drug Delivery: A Review. Curr. Pharm. Des. 2018, 24, 5296–5312. [Google Scholar] [CrossRef]
- De Palma, M.; Mazzieri, R.; Politi, L.S.; Pucci, F.; Zonari, E.; Sitia, G.; Mazzoleni, S.; Moi, D.; Venneri, M.A.; Indraccolo, S. Tumor-targeted interferon-α delivery by Tie2-expressing monocytes inhibits tumor growth and metastasis. Cancer Cell 2008, 14, 299–311. [Google Scholar] [CrossRef] [Green Version]
- He, X.; Cao, H.; Wang, H.; Tan, T.; Yu, H.; Zhang, P.; Yin, Q.; Zhang, Z.; Li, Y. Inflammatory monocytes loading protease-sensitive nanoparticles enable lung metastasis targeting and intelligent drug release for anti-metastasis therapy. Nano Lett. 2017, 17, 5546–5554. [Google Scholar] [CrossRef] [PubMed]
- Sugimoto, S.; Iwasaki, Y. Surface modification of macrophages with nucleic acid aptamers for enhancing the immune response against tumor cells. Bioconjug. Chem. 2018, 29, 4160–4167. [Google Scholar] [CrossRef] [PubMed]
- Holden, C.A.; Yuan, Q.; Yeudall, W.A.; Lebman, D.A.; Yang, H. Surface engineering of macrophages with nanoparticles to generate a cell nanoparticle hybrid vehicle for hypoxia-targeted drug delivery. Int. J. Nanomed. 2010, 5, 25–36. [Google Scholar]
- Cao, H.; Dan, Z.; He, X.; Zhang, Z.; Yu, H.; Yin, Q.; Li, Y. Liposomes coated with isolated macrophage membrane can target lung metastasis of breast cancer. ACS Nano 2016, 10, 7738–7748. [Google Scholar] [CrossRef]
- Molinaro, R.; Corbo, C.; Martinez, J.O.; Taraballi, F.; Evangelopoulos, M.; Minardi, S.; Yazdi, I.K.; Zhao, P.; De Rosa, E.; Sherman, M. Biomimetic proteolipid vesicles for targeting inflamed tissues. Nat. Mater. 2016, 15, 1037–1046. [Google Scholar] [CrossRef]
- Parodi, A.; Quattrocchi, N.; Van De Ven, A.L.; Chiappini, C.; Evangelopoulos, M.; Martinez, J.O.; Brown, B.S.; Khaled, S.Z.; Yazdi, I.K.; Enzo, M.V. Synthetic nanoparticles functionalized with biomimetic leukocyte membranes possess cell-like functions. Nat. Nanotechnol. 2013, 8, 61–68. [Google Scholar] [CrossRef] [Green Version]
- He, H.; Guo, C.; Wang, J.; Korzun, W.J.; Wang, X.-Y.; Ghosh, S.; Yang, H. Leutusome: A biomimetic nanoplatform integrating plasma membrane components of leukocytes and tumor cells for remarkably enhanced solid tumor homing. Nano Lett. 2018, 18, 6164–6174. [Google Scholar] [CrossRef]
- Zeng, R.; Spolski, R.; Finkelstein, S.E.; Oh, S.; Kovanen, P.E.; Hinrichs, C.S.; Pise-Masison, C.A.; Radonovich, M.F.; Brady, J.N.; Restifo, N.P. Synergy of IL-21 and IL-15 in regulating CD8+ T cell expansion and function. J. Exp. Med. 2005, 201, 139–148. [Google Scholar] [CrossRef] [Green Version]
- Thompson, J.A.; Lee, D.J.; Cox, W.W.; Lindgren, C.G.; Collins, C.; Neraas, K.A.; Dennin, R.A.; Fefer, A. Recombinant interleukin 2 toxicity, pharmacokinetics, and immunomodulatory effects in a phase I trial. Cancer Res. 1987, 47, 4202–4207. [Google Scholar]
- Berger, C.; Berger, M.; Hackman, R.C.; Gough, M.; Elliott, C.; Jensen, M.C.; Riddell, S.R. Safety and immunologic effects of IL-15 administration in nonhuman primates. Blood 2009, 114, 2417–2426. [Google Scholar] [CrossRef] [Green Version]
- Stephan, M.T.; Moon, J.J.; Um, S.H.; Bershteyn, A.; Irvine, D.J. Therapeutic cell engineering with surface-conjugated synthetic nanoparticles. Nat. Med. 2010, 16, 1035. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tang, L.; Zheng, Y.; Melo, M.B.; Mabardi, L.; Castaño, A.P.; Xie, Y.-Q.; Li, N.; Kudchodkar, S.B.; Wong, H.C.; Jeng, E.K. Enhancing T cell therapy through TCR-signaling-responsive nanoparticle drug delivery. Nat. Biotechnol. 2018, 36, 707–716. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, B.; Abraham, W.D.; Zheng, Y.; López, S.C.B.; Luo, S.S.; Irvine, D.J. Active targeting of chemotherapy to disseminated tumors using nanoparticle-carrying T cells. Sci. Transl. Med. 2015, 7, 291ra94. [Google Scholar] [CrossRef] [Green Version]
- Liu, L.; Chen, Q.; Ruan, C.; Chen, X.; He, X.; Zhang, Y.; Zhang, Y.; Lu, Y.; Guo, Q.; Zhou, W. Nano-engineered lymphocytes for alleviating suppressive tumor immune microenvironment. Appl. Mater. Today 2019, 16, 273–279. [Google Scholar] [CrossRef]
- Acosta, N.D.; Golub, S.H. The new federalism: State policies regarding embryonic stem cell research. J. Law Med. Ethics 2016, 44, 419–436. [Google Scholar] [CrossRef]
- Cheng, H.; Kastrup, C.J.; Ramanathan, R.; Siegwart, D.J.; Ma, M.; Bogatyrev, S.R.; Xu, Q.; Whitehead, K.A.; Langer, R.; Anderson, D.G. Nanoparticulate cellular patches for cell-mediated tumoritropic delivery. ACS Nano 2010, 4, 625–631. [Google Scholar] [CrossRef] [Green Version]
- Ko, I.K.; Kean, T.J.; Dennis, J.E. Targeting mesenchymal stem cells to activated endothelial cells. Biomaterials 2009, 30, 3702–3710. [Google Scholar] [CrossRef] [Green Version]
- Zhao, W.; Loh, W.; Droujinine, I.A.; Teo, W.; Kumar, N.; Schafer, S.; Cui, C.H.; Zhang, L.; Sarkar, D.; Karnik, R. Mimicking the inflammatory cell adhesion cascade by nucleic acid aptamer programmed cell-cell interactions. FASEB J. 2011, 25, 3045–3056. [Google Scholar] [CrossRef] [Green Version]
- Sarkar, D.; Spencer, J.A.; Phillips, J.A.; Zhao, W.; Schafer, S.; Spelke, D.P.; Mortensen, L.J.; Ruiz, J.P.; Vemula, P.K.; Sridharan, R. Engineered cell homing. Blood 2011, 118, e184–e191. [Google Scholar] [CrossRef]
- Cheng, H.; Byrska-Bishop, M.; Zhang, C.T.; Kastrup, C.J.; Hwang, N.S.; Tai, A.K.; Lee, W.W.; Xu, X.; Nahrendorf, M.; Langer, R. Stem cell membrane engineering for cell rolling using peptide conjugation and tuning of cell selectin interaction kinetics. Biomaterials 2012, 33, 5004–5012. [Google Scholar] [CrossRef] [Green Version]
- Sarkar, D.; Vemula, P.K.; Teo, G.S.; Spelke, D.; Karnik, R.; Wee, L.Y.; Karp, J.M. Chemical engineering of mesenchymal stem cells to induce a cell rolling response. Bioconjug. Chem. 2008, 19, 2105–2109. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez-Rey, E.; Anderson, P.; González, M.A.; Rico, L.; Büscher, D.; Delgado, M. Human adult stem cells derived from adipose tissue protect against experimental colitis and sepsis. Gut 2009, 58, 929–939. [Google Scholar] [CrossRef] [PubMed]
- Ko, I.K.; Kim, B.-G.; Awadallah, A.; Mikulan, J.; Lin, P.; Letterio, J.J.; Dennis, J.E. Targeting improves MSC treatment of inflammatory bowel disease. Mol. Ther. 2010, 18, 1365–1372. [Google Scholar] [CrossRef] [PubMed]
- Lapidot, T.; Dar, A.; Kollet, O. How do stem cells find their way home? Blood 2005, 106, 1901–1910. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sackstein, R.; Merzaban, J.S.; Cain, D.W.; Dagia, N.M.; Spencer, J.A.; Lin, C.P.; Wohlgemuth, R. Ex vivo glycan engineering of CD44 programs human multipotent mesenchymal stromal cell trafficking to bone. Nat. Med. 2008, 14, 181. [Google Scholar] [CrossRef]
- Zhao, W.; Schafer, S.; Choi, J.; Yamanaka, Y.J.; Lombardi, M.L.; Bose, S.; Carlson, A.L.; Phillips, J.A.; Teo, W.; Droujinine, I.A. Cell-surface sensors for real-time probing of cellular environments. Nat. Nanotechnol. 2011, 6, 524–531. [Google Scholar] [CrossRef] [Green Version]
- King, I.; Bermudes, D.; Lin, S.; Belcourt, M.; Pike, J.; Troy, K.; Le, T.; Ittensohn, M.; Mao, J.; Lang, W. Tumor-targeted Salmonella expressing cytosine deaminase as an anticancer agent. Hum. Gene Ther. 2002, 13, 1225–1233. [Google Scholar] [CrossRef] [Green Version]
- Low, K.B.; Ittensohn, M.; Le, T.; Platt, J.; Sodi, S.; Amoss, M.; Ash, O.; Carmichael, E.; Chakraborty, A.; Fischer, J. Lipid A mutant Salmonella with suppressed virulence and TNFα induction retain tumor-targeting in vivo. Nat. Biotechnol. 1999, 17, 37–41. [Google Scholar] [CrossRef]
- Pawelek, J.M.; Low, K.B.; Bermudes, D. Tumor-targeted Salmonella as a novel anticancer vector. Cancer Res. 1997, 57, 4537–4544. [Google Scholar]
- Xiang, S.; Fruehauf, J.; Li, C.J. Short hairpin RNA-expressing bacteria elicit RNA interference in mammals. Nat. Biotechnol. 2006, 24, 697–702. [Google Scholar] [CrossRef]
- Pawelek, J.M.; Low, K.B.; Bermudes, D. Bacteria as tumour-targeting vectors. Lancet Oncol. 2003, 4, 548–556. [Google Scholar] [CrossRef]
- Cao, Z.; Cheng, S.; Wang, X.; Pang, Y.; Liu, J. Camouflaging bacteria by wrapping with cell membranes. Nat. Commun. 2019, 10, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Hjelm, A.; Söderström, B.; Vikström, D.; Jong, W.S.; Luirink, J.; de Gier, J.-W. Autotransporter-based antigen display in bacterial ghosts. Appl. Environ. Microbiol. 2015, 81, 726–735. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kudela, P.; Paukner, S.; Mayr, U.B.; Cholujova, D.; Schwarczova, Z.; Sedlak, J.; Bizik, J.; Lubitz, W. Bacterial ghosts as novel efficient targeting vehicles for DNA delivery to the human monocyte-derived dendritic cells. J. Immunother. 2005, 28, 136–143. [Google Scholar] [CrossRef] [PubMed]
- Paukner, S.; Kudela, P.; Kohl, G.; Schlapp, T.; Friedrichs, S.; Lubitz, W. DNA-loaded bacterial ghosts efficiently mediate reporter gene transfer and expression in macrophages. Mol. Ther. 2005, 11, 215–223. [Google Scholar] [CrossRef] [PubMed]
- Paukner, S.; Kohl, G.; Lubitz, W. Bacterial ghosts as novel advanced drug delivery systems: Antiproliferative activity of loaded doxorubicin in human Caco-2 cells. J. Control. Release 2004, 94, 63–74. [Google Scholar] [CrossRef]
- Kudela, P.; Koller, V.J.; Lubitz, W. Bacterial ghosts (BGs)—Advanced antigen and drug delivery system. Vaccine 2010, 28, 5760–5767. [Google Scholar] [CrossRef]
- Lubitz, P.; Mayr, U.B.; Lubitz, W. Applications of bacterial ghosts in biomedicine. In Pharmaceutical Biotechnology; Guzmán, C.A., Feuerstein, G.Z., Eds.; Springer: New York, NY, USA, 2009; pp. 159–170. [Google Scholar]
- Gao, W.; Fang, R.H.; Thamphiwatana, S.; Luk, B.T.; Li, J.; Angsantikul, P.; Zhang, Q.; Hu, C.-M.J.; Zhang, L. Modulating antibacterial immunity via bacterial membrane-coated nanoparticles. Nano Lett. 2015, 15, 1403–1409. [Google Scholar] [CrossRef] [Green Version]
- Gujrati, V.; Kim, S.; Kim, S.-H.; Min, J.J.; Choy, H.E.; Kim, S.C.; Jon, S. Bioengineered bacterial outer membrane vesicles as cell-specific drug-delivery vehicles for cancer therapy. ACS Nano 2014, 8, 1525–1537. [Google Scholar] [CrossRef]
- Sagnella, S.M.; Trieu, J.; Brahmbhatt, H.; MacDiarmid, J.A.; MacMillan, A.; Whan, R.M.; Fife, C.M.; McCarroll, J.A.; Gifford, A.J.; Ziegler, D.S. Targeted doxorubicin-loaded bacterially derived nano-cells for the treatment of neuroblastoma. Mol. Cancer Ther. 2018, 17, 1012–1023. [Google Scholar] [CrossRef] [Green Version]
- MacDiarmid, J.A.; Mugridge, N.B.; Weiss, J.C.; Phillips, L.; Burn, A.L.; Paulin, R.P.; Haasdyk, J.E.; Dickson, K.-A.; Brahmbhatt, V.N.; Pattison, S.T. Bacterially derived 400 nm particles for encapsulation and cancer cell targeting of chemotherapeutics. Cancer Cell 2007, 11, 431–445. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- György, B.; Hung, M.E.; Breakefield, X.O.; Leonard, J.N. Therapeutic applications of extracellular vesicles: Clinical promise and open questions. Annu. Rev. Pharmacol. Toxicol. 2015, 55, 439–464. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tian, T.; Zhang, H.-X.; He, C.-P.; Fan, S.; Zhu, Y.-L.; Qi, C.; Huang, N.-P.; Xiao, Z.-D.; Lu, Z.-H.; Tannous, B.A. Surface functionalized exosomes as targeted drug delivery vehicles for cerebral ischemia therapy. Biomaterials 2018, 150, 137–149. [Google Scholar] [CrossRef] [PubMed]
- Sun, D.; Zhuang, X.; Xiang, X.; Liu, Y.; Zhang, S.; Liu, C.; Barnes, S.; Grizzle, W.; Miller, D.; Zhang, H.-G. A novel nanoparticle drug delivery system: The anti-inflammatory activity of curcumin is enhanced when encapsulated in exosomes. Mol. Ther. 2010, 18, 1606–1614. [Google Scholar] [CrossRef]
- Iero, M.; Valenti, R.; Huber, V.; Filipazzi, P.; Parmiani, G.; Fais, S.; Rivoltini, L. Tumour-released exosomes and their implications in cancer immunity. Cell Death Differ. 2008, 15, 80–88. [Google Scholar] [CrossRef]
- Skog, J.; Würdinger, T.; Van Rijn, S.; Meijer, D.H.; Gainche, L.; Curry, W.T., Jr.; Carter, B.S.; Krichevsky, A.M.; Breakefield, X.O. Glioblastoma microvesicles transport RNA and proteins that promote tumour growth and provide diagnostic biomarkers. Nat. Cell Biol. 2008, 10, 1470–1476. [Google Scholar] [CrossRef]
- Alvarez-Erviti, L.; Seow, Y.; Yin, H.; Betts, C.; Lakhal, S.; Wood, M.J. Delivery of siRNA to the mouse brain by systemic injection of targeted exosomes. Nat. Biotechnol. 2011, 29, 341–345. [Google Scholar] [CrossRef]
- Kamerkar, S.; LeBleu, V.S.; Sugimoto, H.; Yang, S.; Ruivo, C.F.; Melo, S.A.; Lee, J.J.; Kalluri, R. Exosomes facilitate therapeutic targeting of oncogenic KRAS in pancreatic cancer. Nature 2017, 546, 498–503. [Google Scholar] [CrossRef]
- Ohno, S.-I.; Takanashi, M.; Sudo, K.; Ueda, S.; Ishikawa, A.; Matsuyama, N.; Fujita, K.; Mizutani, T.; Ohgi, T.; Ochiya, T. Systemically injected exosomes targeted to EGFR deliver antitumor microRNA to breast cancer cells. Mol. Ther. 2013, 21, 185–191. [Google Scholar] [CrossRef] [Green Version]
- Lai, C.P.; Mardini, O.; Ericsson, M.; Prabhakar, S.; Maguire, C.A.; Chen, J.W.; Tannous, B.A.; Breakefield, X.O. Dynamic biodistribution of extracellular vesicles in vivo using a multimodal imaging reporter. ACS Nano 2014, 8, 483–494. [Google Scholar] [CrossRef] [Green Version]
- Kooijmans, S.A.; Schiffelers, R.M.; Zarovni, N.; Vago, R. Modulation of tissue tropism and biological activity of exosomes and other extracellular vesicles: New nanotools for cancer treatment. Pharmacol. Res. 2016, 111, 487–500. [Google Scholar] [CrossRef] [PubMed]
- Kooijmans, S.; Fliervoet, L.; Van Der Meel, R.; Fens, M.; Heijnen, H.; en Henegouwen, P.V.B.; Vader, P.; Schiffelers, R. PEGylated and targeted extracellular vesicles display enhanced cell specificity and circulation time. J. Control. Release 2016, 224, 77–85. [Google Scholar] [CrossRef] [PubMed]
- Nakase, I.; Futaki, S. Combined treatment with a pH-sensitive fusogenic peptide and cationic lipids achieves enhanced cytosolic delivery of exosomes. Sci. Rep. 2015, 5, 10112. [Google Scholar] [CrossRef] [PubMed]
- Qi, H.; Liu, C.; Long, L.; Ren, Y.; Zhang, S.; Chang, X.; Qian, X.; Jia, H.; Zhao, J.; Sun, J. Blood exosomes endowed with magnetic and targeting properties for cancer therapy. ACS Nano 2016, 10, 3323–3333. [Google Scholar] [CrossRef] [PubMed]
- Smyth, T.; Petrova, K.; Payton, N.M.; Persaud, I.; Redzic, J.S.; Graner, M.W.; Smith-Jones, P.; Anchordoquy, T.J. Surface functionalization of exosomes using click chemistry. Bioconjug. Chem. 2014, 25, 1777–1784. [Google Scholar] [CrossRef] [Green Version]
- Wan, Y.; Wang, L.; Zhu, C.; Zheng, Q.; Wang, G.; Tong, J.; Fang, Y.; Xia, Y.; Cheng, G.; He, X. Aptamer-conjugated extracellular nanovesicles for targeted drug delivery. Cancer Res. 2018, 78, 798–808. [Google Scholar] [CrossRef] [Green Version]
- He, H.; Liu, L.; Morin, E.E.; Liu, M.; Schwendeman, A. Survey of Clinical Translation of Cancer Nanomedicines—Lessons Learned from Successes and Failures. Acc. Chem. Res. 2019, 52, 2445–2461. [Google Scholar] [CrossRef]






© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liu, L.; He, H.; Liu, J. Advances on Non-Genetic Cell Membrane Engineering for Biomedical Applications. Polymers 2019, 11, 2017. https://doi.org/10.3390/polym11122017
Liu L, He H, Liu J. Advances on Non-Genetic Cell Membrane Engineering for Biomedical Applications. Polymers. 2019; 11(12):2017. https://doi.org/10.3390/polym11122017
Chicago/Turabian StyleLiu, Lisha, Hongliang He, and Jianping Liu. 2019. "Advances on Non-Genetic Cell Membrane Engineering for Biomedical Applications" Polymers 11, no. 12: 2017. https://doi.org/10.3390/polym11122017