1. Introduction
The “traditional” plastics derived from coal, natural gas, and oil are one of the most environmentally harmful substances produced by humans, but they are also an important and useful part of our society [
1]. From mobile phones to packaging, it is difficult to imagine a world without them. Plastics are extremely versatile; they can be transparent, cost-effective, lightweight, strong, and durable, and they also possess vital properties that make them useful in medical, agricultural, domestic, and industrial applications [
2]. While these properties are desirable, the highly stable structure of plastics also makes the process of natural degradation difficult; therefore, these materials are overused, and they persist in soil, marine, and freshwater ecosystems when they are irresponsibly dumped [
3,
4]. Even attempting to destroy plastics via incineration can cause major air pollution by releasing toxins, like dioxins, furans, mercury, and polychlorinated biphenyls [
1]. Polyhydroxyalkanoates (PHAs) are a family of polyhydroxyesters with 3-, 4-, 5- and 6-hydroxyalkanoic acids that are biocompatible, biodegradable, and non-toxic organic polyesters synthesized by certain bacteria and plants from renewable sources [
5,
6]. PHAs function as a storage reservoir of carbon that can be accessed whenever required by the microorganism, often in the form of intercellular granules [
6]. Currently, PHAs are being used either as a blend or in its pure form in a variety of applications; scl-PHAs (short chain length) are predominantly used to produce food packaging and disposable items, and mcl-PHAs (medium chain length-characterized as elastomers) are appropriate for high value-added applications, such as surgical stitching, implants, and drug delivery systems [
5,
7,
8]. There is also the opportunity to functionalize PHB chemically and/or enzymatically (as discussed later), which is among one of the most impervious polymers to be processed in certain plastic items [
9]. Global use and industrialization of PHAs is limited due to high production costs, which results in higher prices than petrol-based polymers, such as polypropylene (PP) or polyethylene (PE), that have a price of approximately US
$0.60–0.87/lb [
10,
11]. PHAs, on the other hand, have costs up to four times greater, between US
$2.25–2.75/lb [
10,
11]. Some businesses have industrialized the production of PHAs, but problems with the purity of substrates (like glucose), the issue that some carbon sources take from human food supplies, and the large amounts of solvents required for the extraction stages makes large scale synthesis difficult [
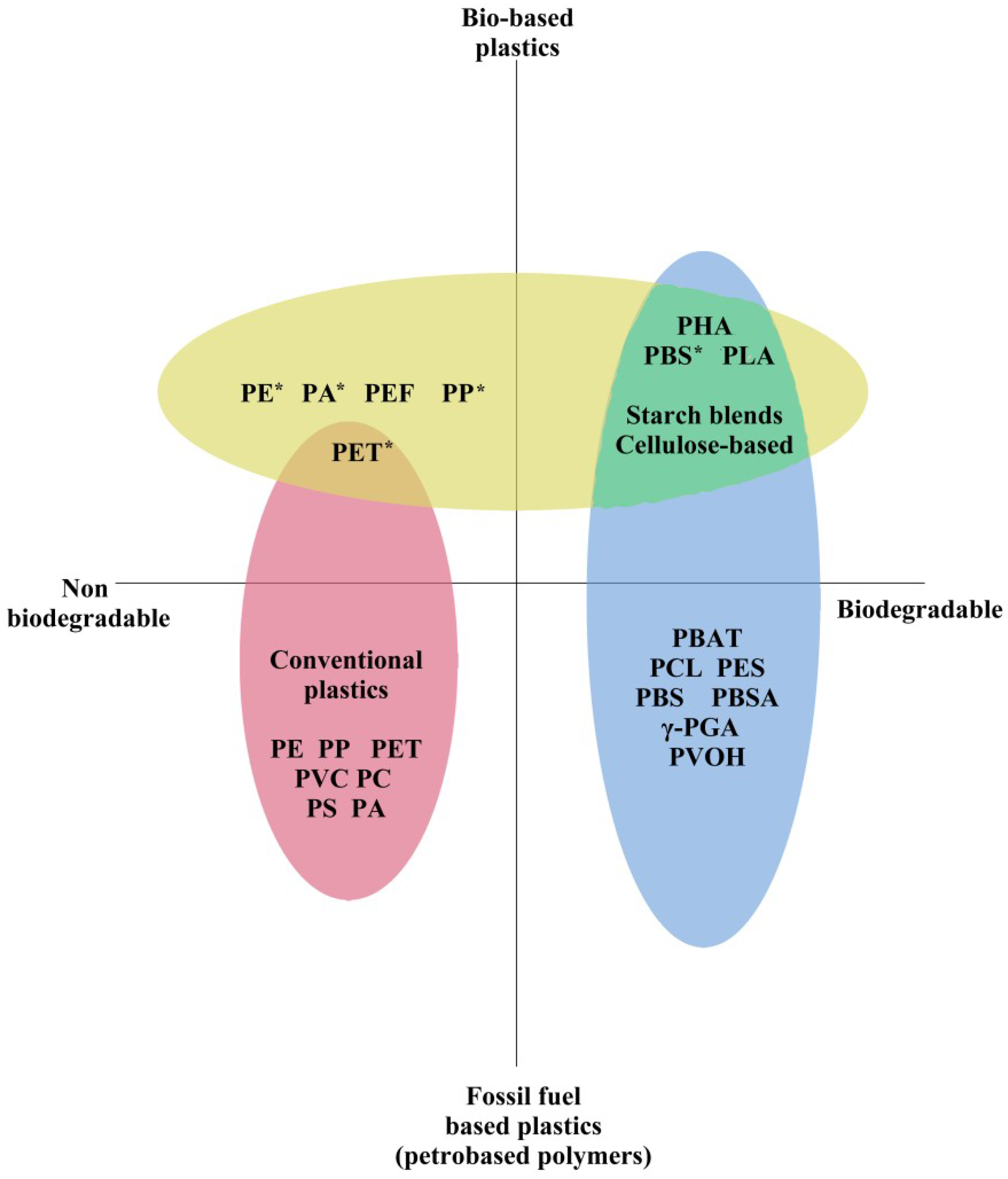
12]. There are a wide range of microbes capable of producing intracellular or extracellular depolymerise enzymes for PHAs, which means it is readily degradable and environmentally safe, as opposed to many other plastics, including full carbon backbone mass polymers, like polystyrene (PS), PP, PE, and others (
Figure 1) [
6,
13]. Besides those polymers featured in
Figure 1, there is a growing market of bio-based building blocks used as precursors for bioplastic production, and of these, succinic acid, 1,4-butanediol, and monoethylene glycol are the most promising [
14].
PS (C
8H
8)
n is fabricated by polymerizing styrene, a building-block chemical used in the manufacture of many products [
15]. It is an extremely versatile plastic that is structurally stable and is often used in products, such as CD cases, and drinks and food packaging. Small amounts of styrene can remain in PS after manufacture; however, the Food and Drug Administration (FDA) has evaluated both the safety of PS and the migrating styrene molecules and found that they are both safe for use. When PS is combined with additives, colorants, or other plastics, it can also be used to make electrical devices, plant pots, or even toys [
15]. When PS is made into a foam material, expanded PS (EPS), or extruded PS (XPS), it can be used for cushions or insulation [
15]. PS has a resin identification code of 6, therefore, it is recyclable, although its biodegradation can take hundreds of years and it is also relatively resistant to photo-oxidation [
16]. As mentioned, PS is widely used, and now there is an abundance of it in outdoor environments. This includes coastal areas, especially in its foam form, and increasingly in the oceans as styrene monomers (SM), styrene dimers (SD), and styrene trimers (ST). PS poses a significant hazard to marine life in its high molar mass form through entanglement and smothering, and in its low molar mass form, potentially through bioaccumulation [
16]. Consequently, PS appears in the red non-biodegradable section of
Figure 1 with other fossil fuel based plastics, meaning PS production leaves a large carbon footprint. Moreover, due to the short lifespan of packaging, a large proportion of PS ends up in landfills, and approximately 13–20% of the solid waste produced is composed of plastic [
17]. Although some European Union (EU) members have banned using landfills, approximately 50% of plastic waste still ends up in one [
18]. In developing countries, plastic consumption and pollution (often marine) is more serious, and it is also less regulated; plastic usage there is believed to be greater than the world average due to more rapid rates of economic growth and urbanization [
18,
19,
20].
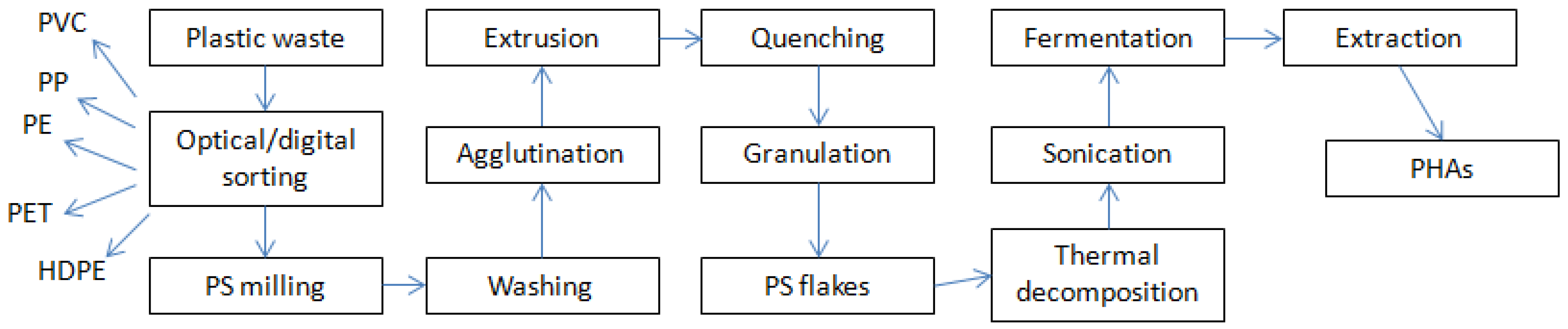
Life Cycle Assessment (LCA) studies have shown that the mechanical recycling of plastic materials is more favorable than other management procedures in relation to overall energy use and emissions contributing to global warming [
21]. The potential pathway of this study’s overall process is summarized in
Figure 2. The quality of waste plastic materials for use in processes such as this one relies on the degree of degradation, the degree of mixing (whether there are impurities or not), and the presence of low molecular weight compounds (additives, contaminants, and degradation products) [
22,
23,
24].
The mechanical recycling of styrene polymers, such as high impact polystyrene (HIPS), has increased over the last decade due to the European Directives on Waste of Electric and Electronic Equipment (WEEE) and End-of-Life Vehicles (ELV) as styrenic plastics are used in their manufacture [
23]. HIPS are a multiphase system with a PS rigid matrix and dispersed rubber particles (polybutadiene) that enhances the blend’s impact properties [
23]. For environmental, social, and economic reasons, innovative and diverse strategies need to be engaged with to encourage the use of alternative biobased plastics that can equal, or even surpass, conventional mass plastics. Waste general purpose PS (GPPS) is a carbon source that could be used in this context to provide an avenue for value-added biopolymer synthesis through fermentation. The utilization of waste PS in this manner would also be in line with the recent global effort to transform Europe’s (and eventually the world’s) economy into a more sustainable one [
25]. In fact, to implement the Circular Economy Action Plan, in January 2018, the European Commission endorsed a new set of measures that includes plans to modify the way plastics are designed, manufactured, applied, and recycled. They also intend that by 2030, all plastic packaging should be recyclable [
25].
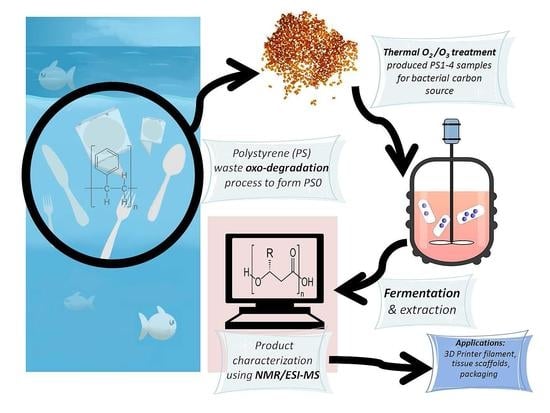
We propose that the fermentation of prodegraded PS (
Figure 2) could be one application of a fraction of global plastic solid waste (PSW) using oxidative degradation as promoted by specific microbial strains [
26]. The prodegraded PS had an acid number of 22 (see
Section 3.1), indicating the presence of carboxylic groups in addition to ketone, acid, or aldehyde C=O groups [
5,
27]. From previous studies, the hydrophilicity of samples can be increased using thermal treatment to increase the acid number, followed by ultrasound sonication to reduce the average molecular mass to stimulate bacterial metabolism [
5]. For alkane metabolism (aerobic), the most common pathway is the oxidation of the terminal methyl group into a carboxylic acid through an alcohol intermediate, which is succeeded by complete mineralization via the β-oxidation pathway [
5,
28,
29]. The degradation of styrene has been reported previously in
Pseudomonas putida CA-3, where epoxidation of styrene occurs, followed by isomerization of the epoxide to form phenylacetaldehyde, which is then oxidized to phenylacetic acid and converted to phenylacetylcoenzyme A (CoA) before further oxidization to acetyl-CoA [
30]. The novelty of this study is that prodegraded PS is the carbon source used, and NMR has been tasked to analyze the thermal degradation process in detail, and, therefore, show the viability of PS as a carbon source with fermentation analysis and polymer sequence characterization. The bacterial strain,
Cupriavidus necator H16 (formerly
Ralstonia eutropha), was selected for this study as it is a reliable producer of PHAs, grows well at relatively low temperatures, has limited immunity to some heavy metals, and has a stable and well-documented genetic profile [
5,
31,
32,
33]. After the PHAs are generated, they are stored as granules, sometimes referred to as “carbonosomes” [
31,
32,
33,
34]. These microbes are known to effectively synthesize PHAs (up to 85% PHA per dry cell weight) under famine and environmentally harsh conditions using a wide range of carbon sources, including: Fatty acids, crude glycerol, hemicellulose, liquefied wood, and methane gas [
34,
35,
36,
37,
38]. The removal of PHAs from biomass uses lyophilization and hot solvent extraction, followed by precipitation in hexane [
5,
33,
35].
In summary, this study details the molecular level structural analysis of PHAs synthesized using C. necator as fed by prodegraded PS (PS0) waste and pretreated samples with thermal oxidation processes (PS1 to 4), as additional carbon sources in nitrogen rich tryptone soya broth (TSB) growth media and nitrogen limited, basal salts media (BSM) during optimal fermentation periods.
4. Discussion
The direct recycling of postconsumer plastics, such as PS, would lead to products of poor quality, with a loss of properties due to aging during the service-life and any thermo-mechanical degradation during reprocessing [
52]. By using prodegraded PS as a carbon source to produce biodegradable plastics, waste PS gains an extended usefulness time-span, rather than becoming a hazardous material. The results of
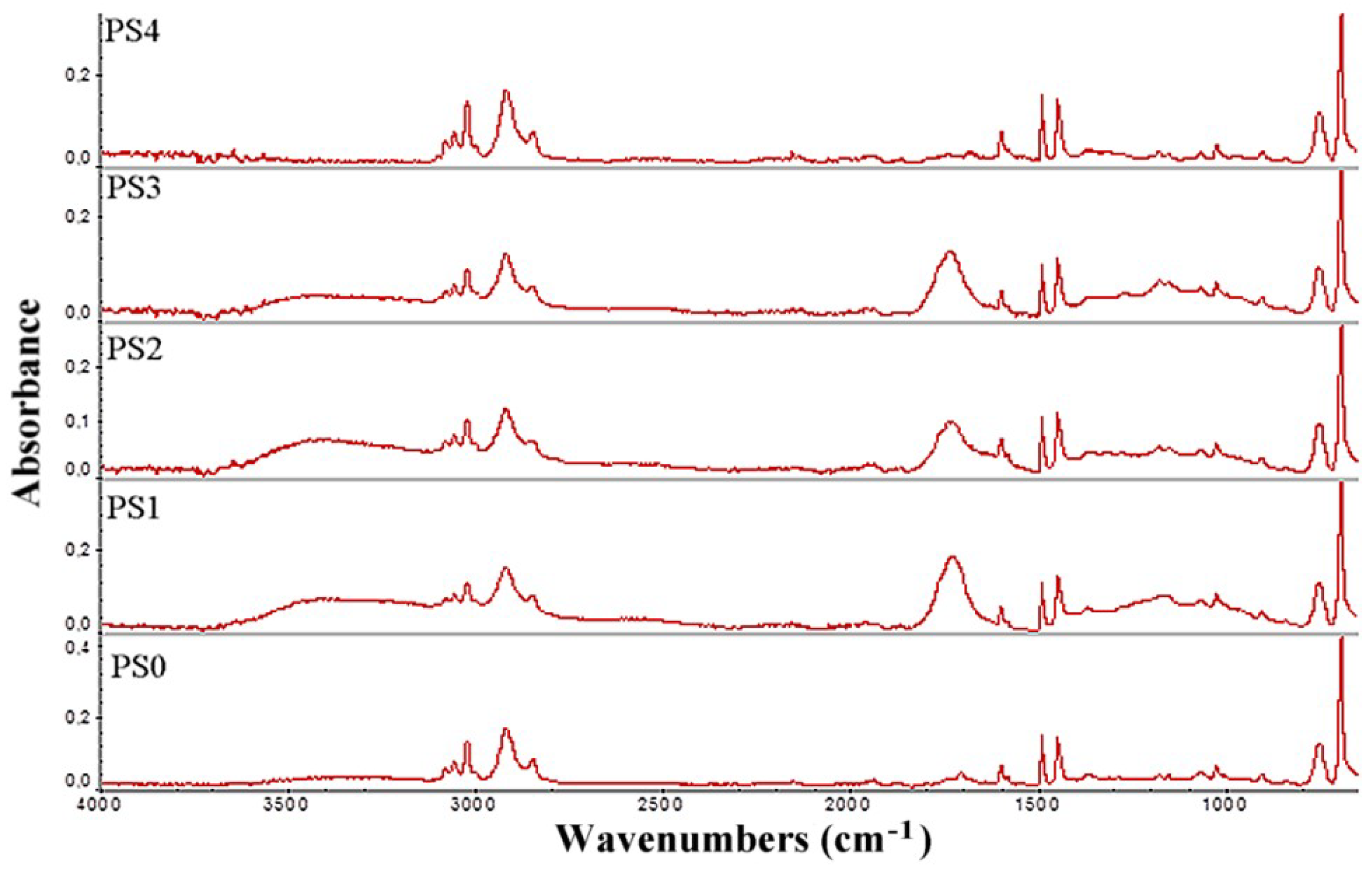
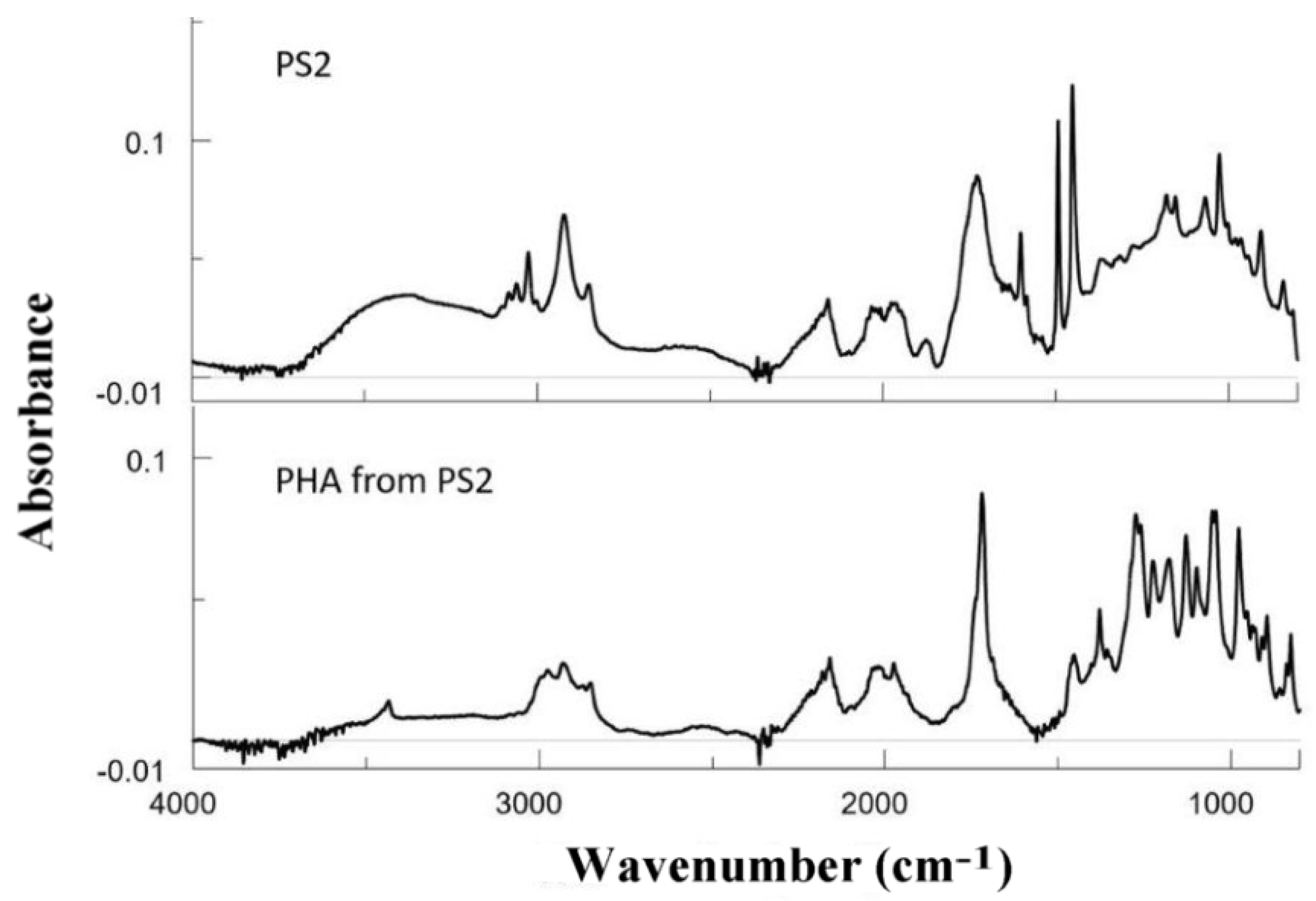
Table 1 and the FTIR data (
Figure 3) clearly show that the thermal pretreatment with ozone increased the presence of ketone or aldehyde groups, further increasing the acid number of samples PS1–3. A treatment time of 20 h at 60 °C was even more effective than the treatment at 80–100 °C for the same period and when 120 °C was used for 20 h the samples are rendered useless for bacterial fermentation.
The GPC results showed a progressive decrease in the average molecular mass of the PS samples when heat exposure was extended. This was expected, however, it was not known that PS4 samples, although possessing the lowest molecular mass, would produce the least biomass and no PHAs after 48 h fermentation. The dispersity (
Ð) between the samples was mostly consistent, apart from PS4 (
Ð = 3.8), possibly showing a higher range of polymers in the sample that could have been produced by the thermal treatment itself. As there was very little growth and no product, these particles could be hazardous so further analysis, or simply avoiding the more extreme conditions used to produce the PS4 samples, is suggested if this kind of pretreatment is utilized by laboratories in the future. It is known that the presence of mixed polymeric fractions, such as impurities in recycled polymeric materials, can affect the macroscopic performance of recyclates [
23]. By using thermal analysis as part of the characterization analysis, it has been observed that the PS fragments were able to pass through the extraction into the PHA because PS readily dissolves in chloroform. If this process was to be adopted in an industrial setting, further filtration steps would have to be added.
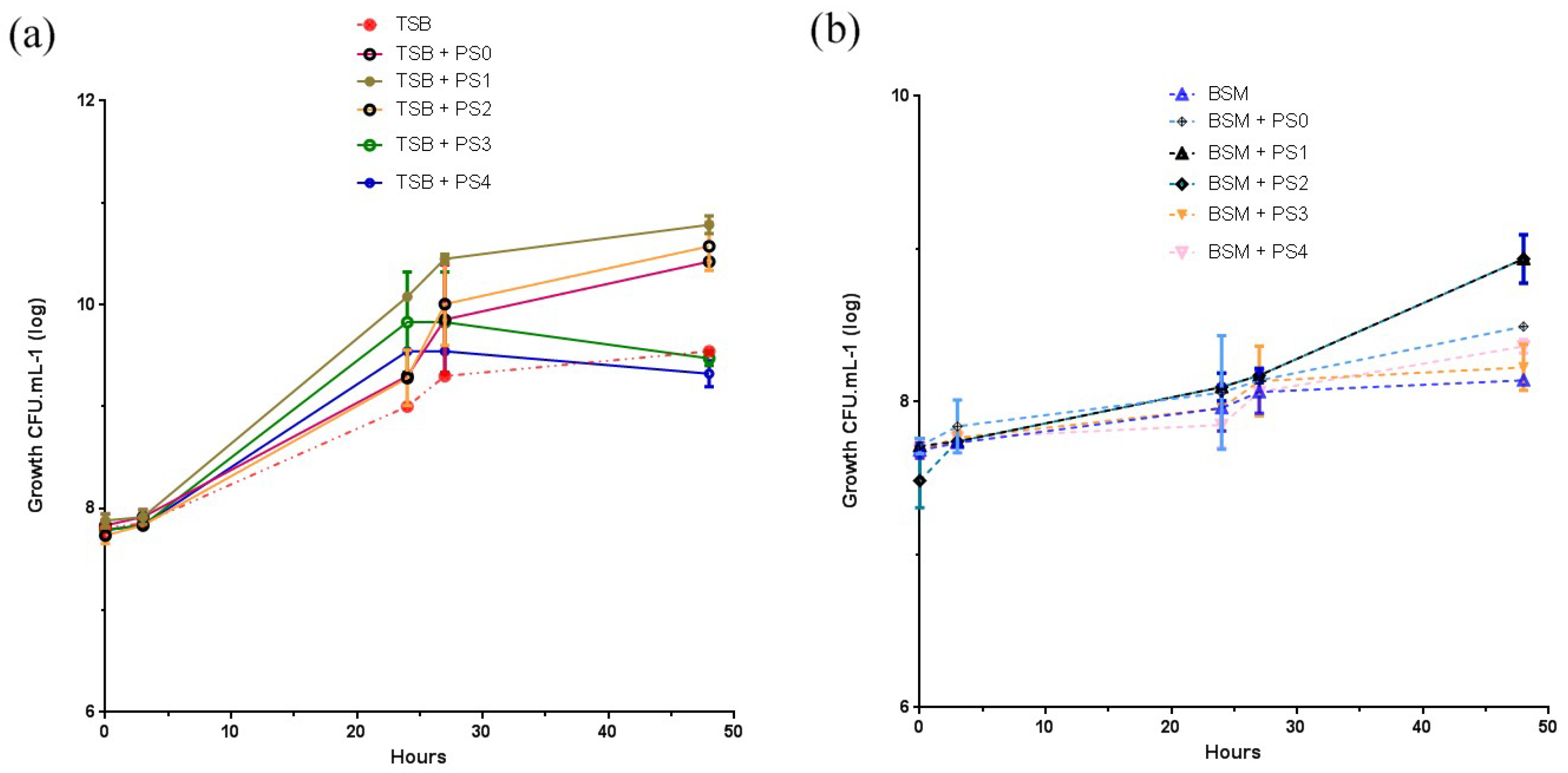
From these results in
Section 3.6, it does appear that the treated PS samples significantly increased the biomass and percentage yield of the PHA. Comparisons of PS1, PS2, and PS3 (the successfully oxidized PS specimens) against BSM showed the greatest significance (
P = 0.001), with PS1 showing a 48% yield. The combination of increased hydrophilicity and reduced average molecular mass undoubtedly contributed to the better emulsification and absorption of the PS carbon chains by the bacteria into the β
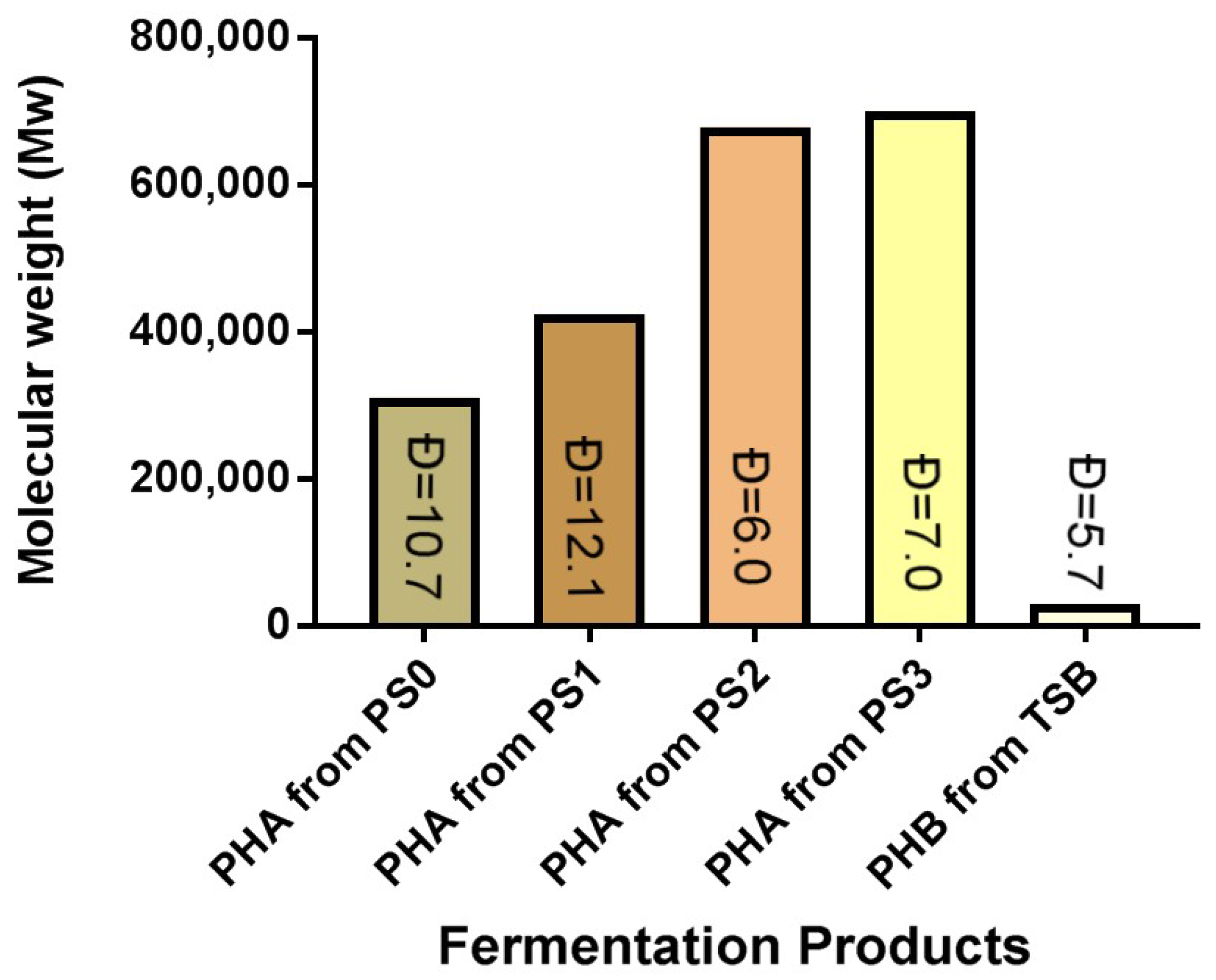
-oxidation pathway, producing PHBV rather than PHB as in the TSB only controls. PS1 sample fermentations also produced relatively low average molecular mass PHAs of 35,000 Da, with the highest distribution value of 12.1 (
Mw/
Mn), compared to PS2 (113,000 Da with
Ð = 6.0) and PS3 (100,000 Da with
Ð = 7.0). When these data are compared to the
Mn results for polyesters synthesized using TSB only, the PHAs synthesized showed a
Mn of 52,000 Da with a distribution of 5.7 (
Mw/
Mn) [
5]. This shows that PS1 samples had the greatest dispersity value, and more varied ranges of polyesters were generated in all prodegraded sample fermentations, but PS1 was the only pretreated sample to give a dispersity value greater than the original prodegraded (PS0) specimen. Again, this is likely due to the higher AN value.
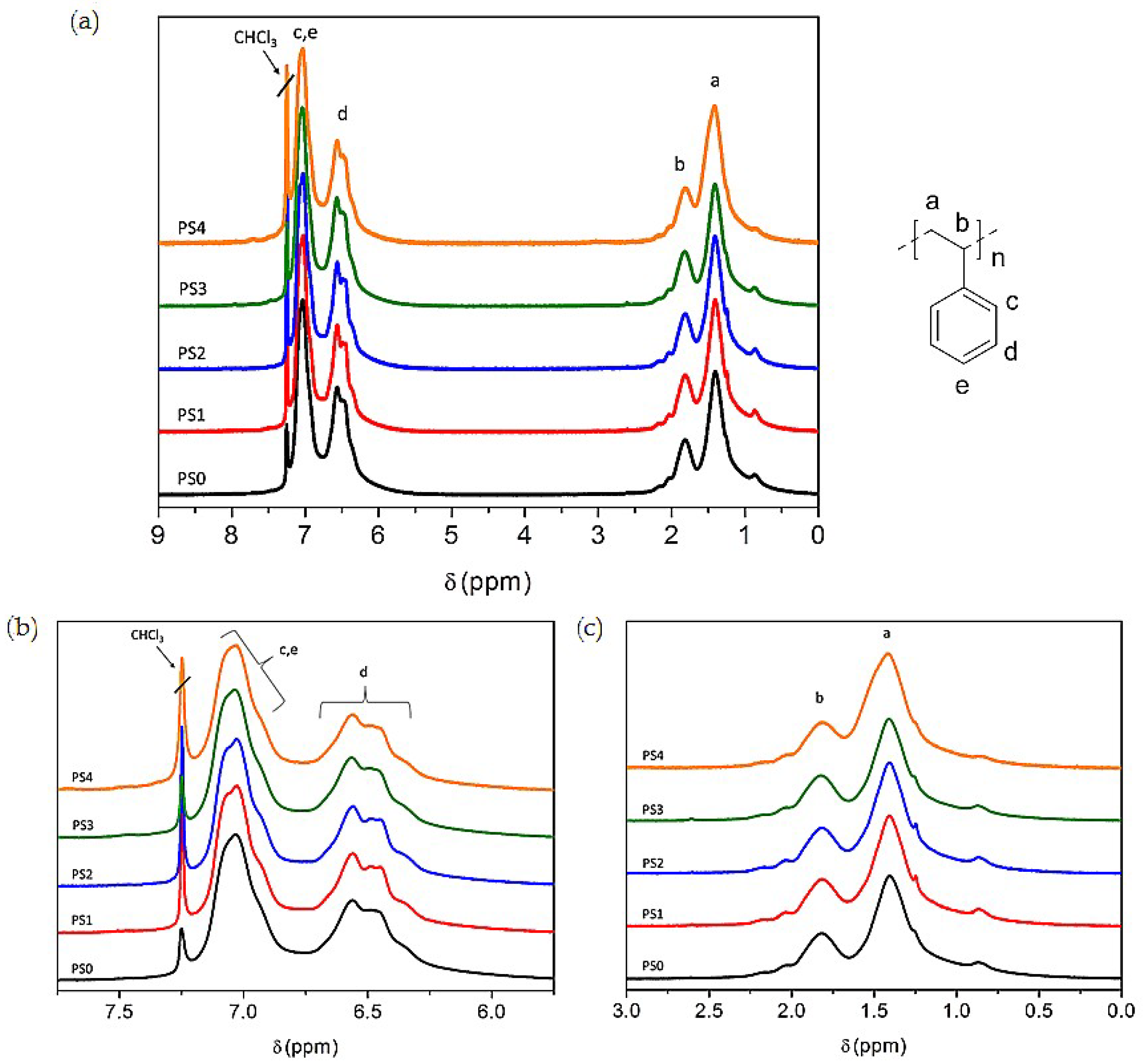
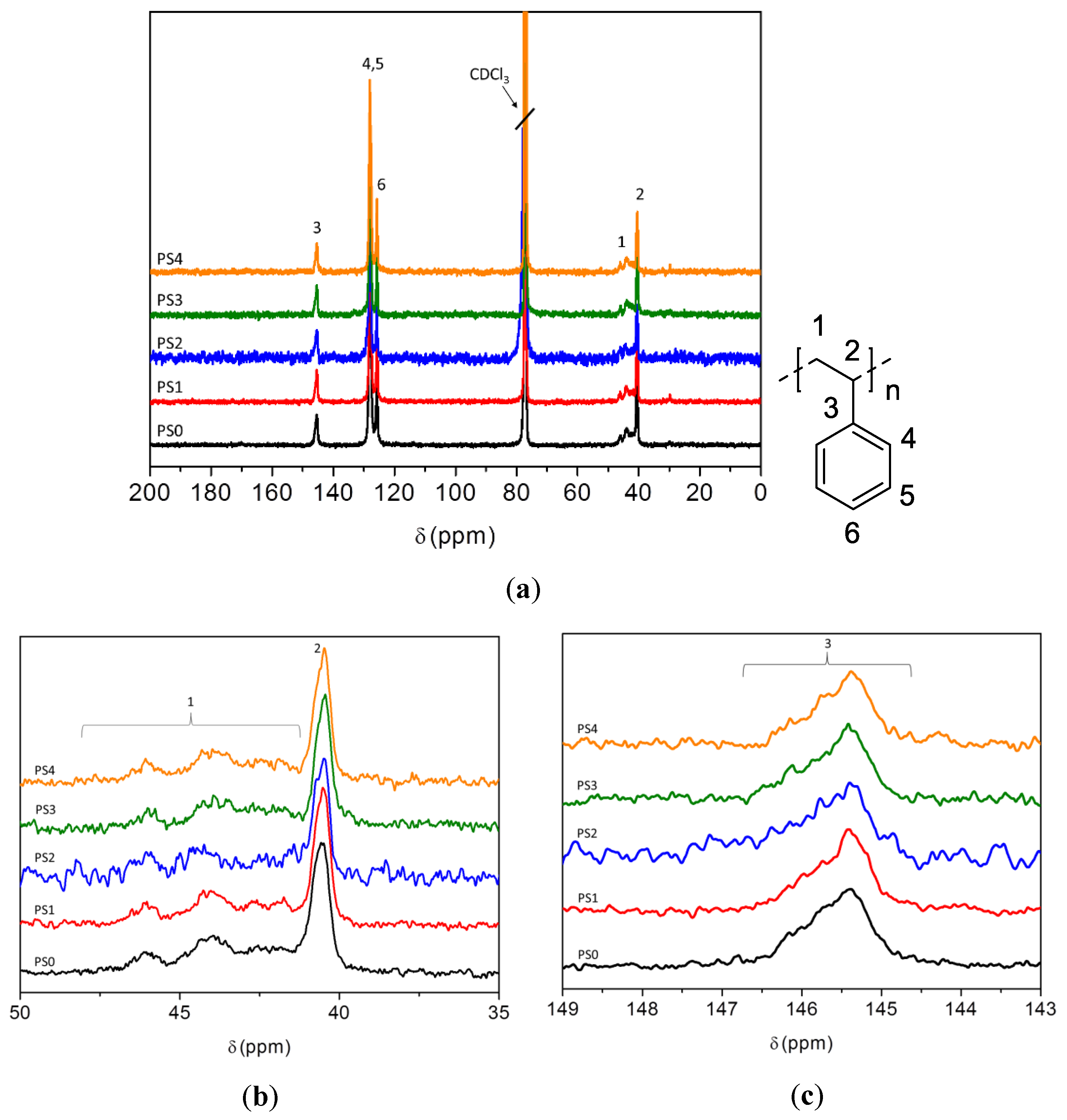
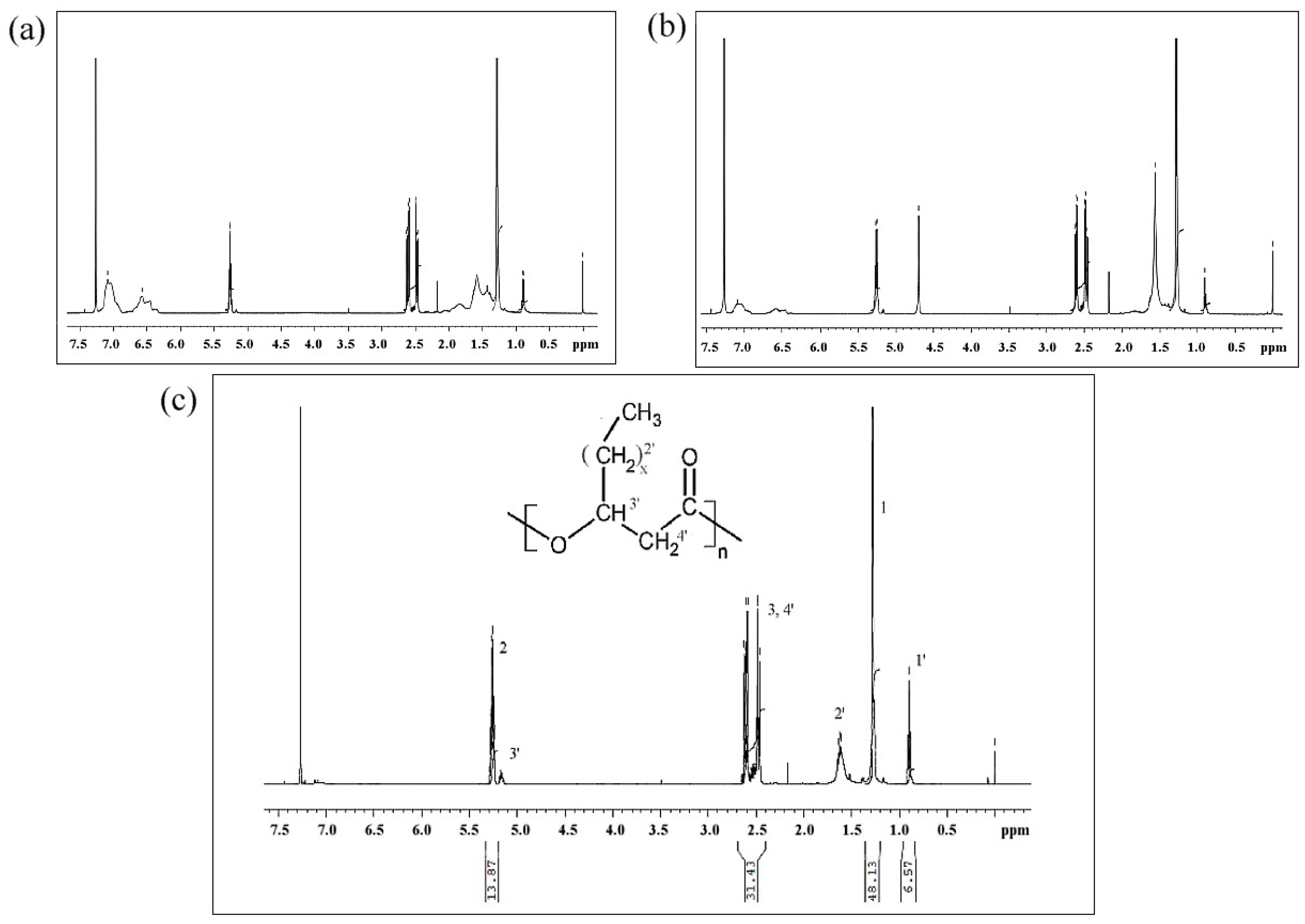
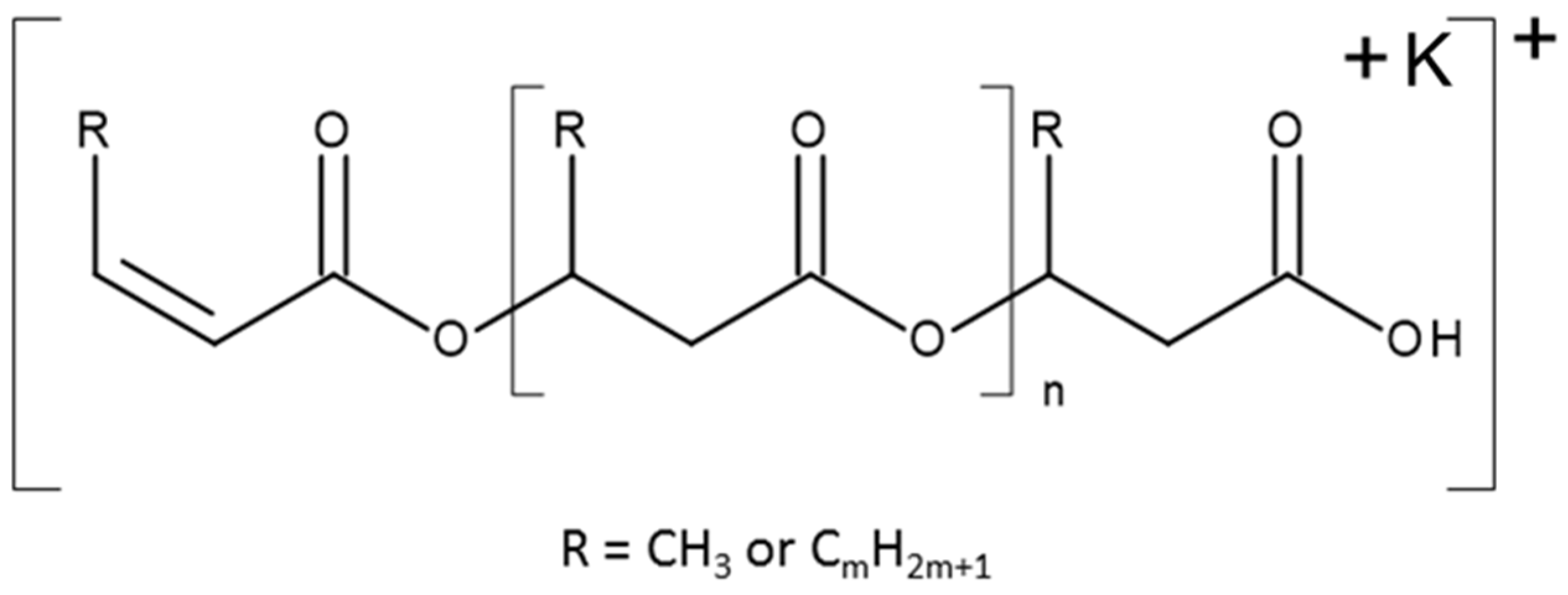
1H-NMR was used to determine the molecular structure of the copolymers produced using fermentation. The 1H-NMR spectra of PSx-PHA revealed the presence of characteristic signals corresponding to the protons of 3-hydroxybutyrate (3-HB) and 3-hydroxyvalerate (3-HV) repeating units. It was found that PHB was produced by bacteria in the TSB only fermentations. After integrating these signals, the content of monomer units other than 3-hydroxybutyrate (3-HB) has been estimated at 12 mol % 3-HV.
ESI-MS determined the molecular structure of the copolymers that were included in the PHAs produced from the prodegraded PS waste samples. This method is known to provide more detail than
1H-NMR or GC when it is applied for PHA characterization. This is because it can corroborate whether a copolymer is randomly distributed or a block copolymer [
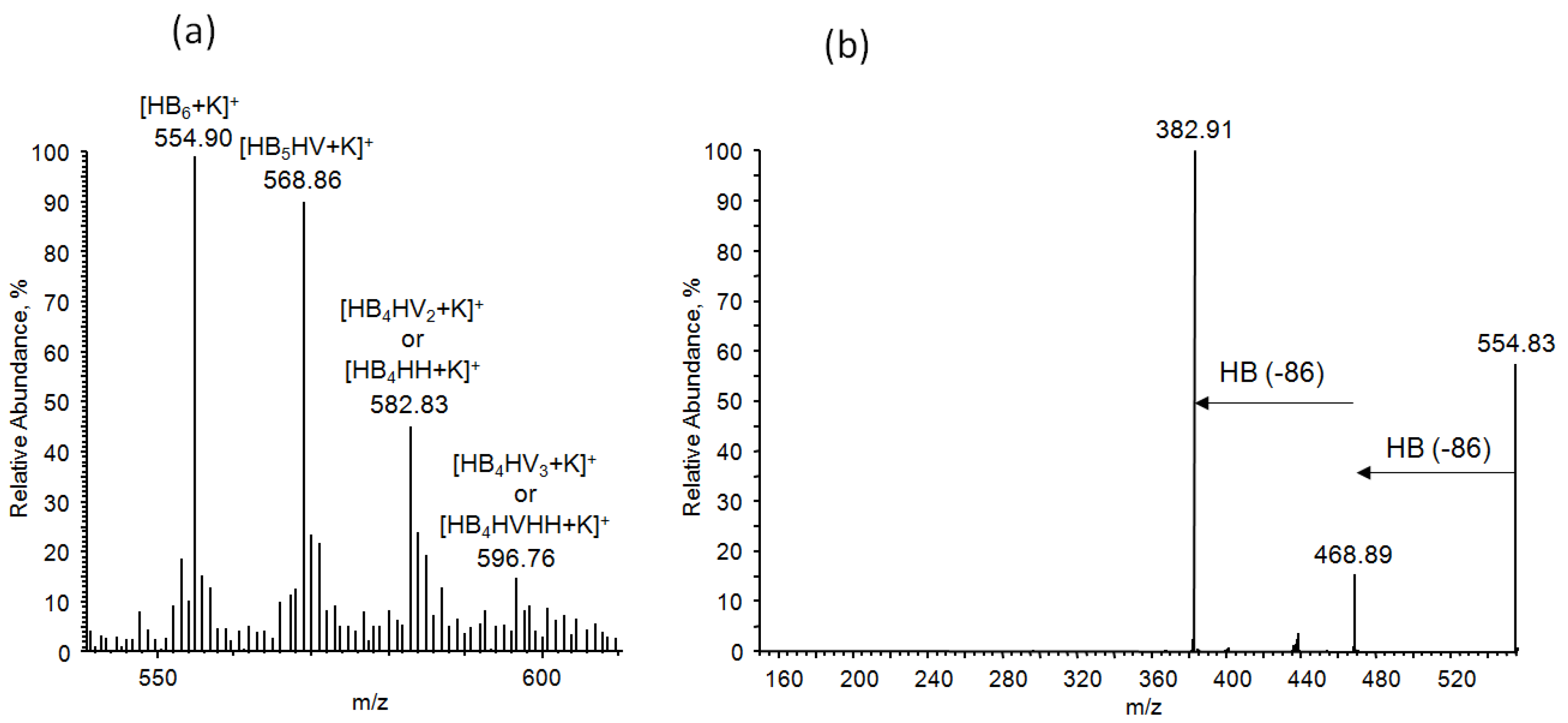
53]. Therefore, it enables the precise sequence analysis of oligomer distribution using the fragmentation technique. The ESI-MS/MS product ions’ spectrum for the selected potassium adducts of the oligomer, [HB
6 + K]
+, at
m/z 555 shown in
Figure 10b confirms that the most intensive ions in the clusters correspond to potassium adducts of 3-hydroxybutyrate oligomers. In
Figure 11, the ESI-MS/MS spectrum for the selected ion, [HB
5 HV + K]
+, at
m/z 569 is presented. During the fragmentation, the breakage of ester bonds along the oligomer chains led to the formation of two series of product ions at
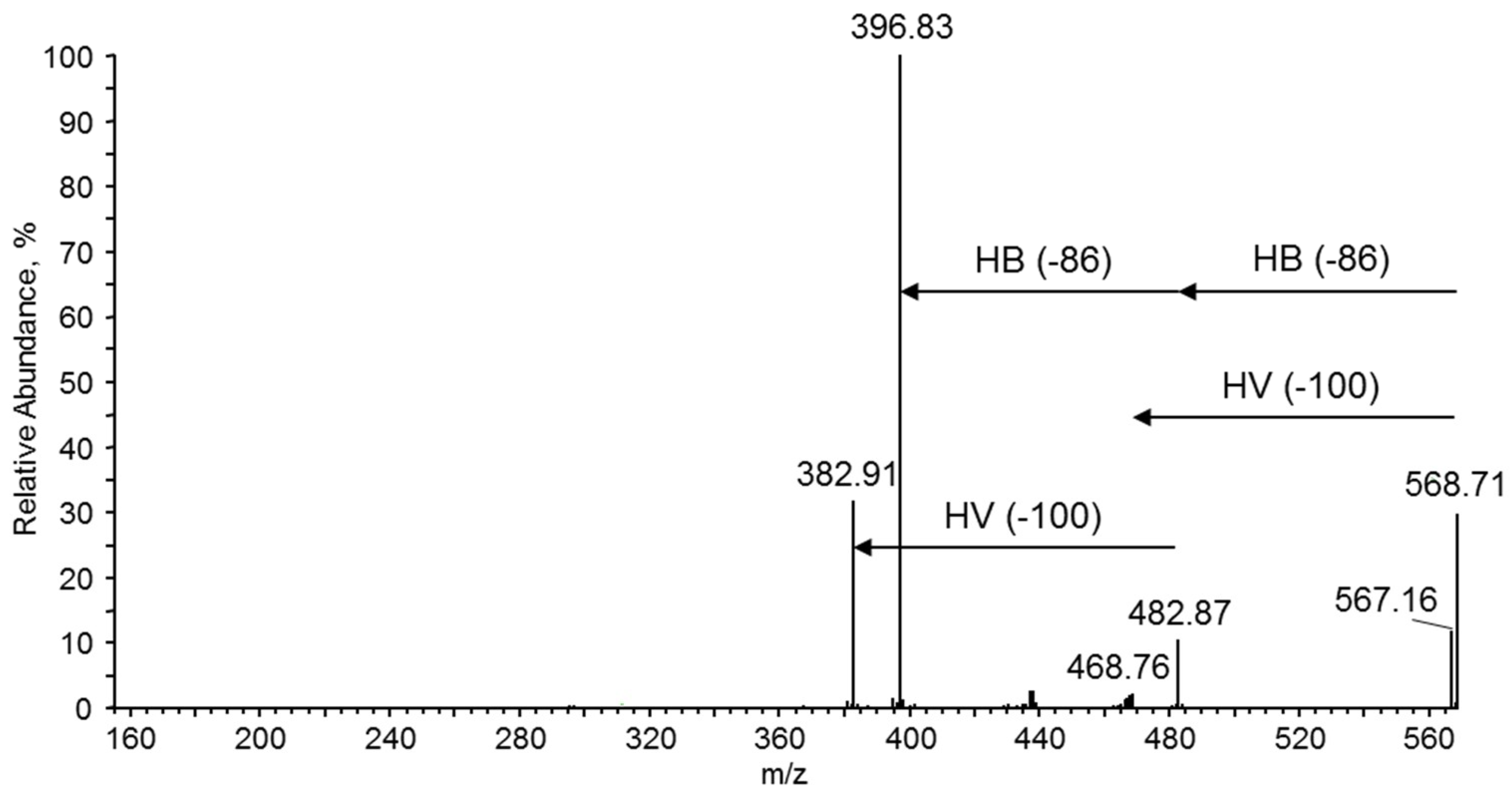
m/z 483, 397 and at
m/z 469, 383. Thus, the product ion at
m/z 483 corresponded to the oligomer formed by the loss of crotonic acid (86 Da) while the product ion at
m/z 469 was formed due to the loss of a valeric acid (100 Da) unit, as seen from the fragmentation pathway shown in
Figure 11. Therefore, the fragmentation spectrum acquired for the precursor ion at
m/z 569 confirms the presence of 3-hydroxybutyrate and the 3-hydroxyvalerate co-monomer units randomly distributed along the polyester chains.
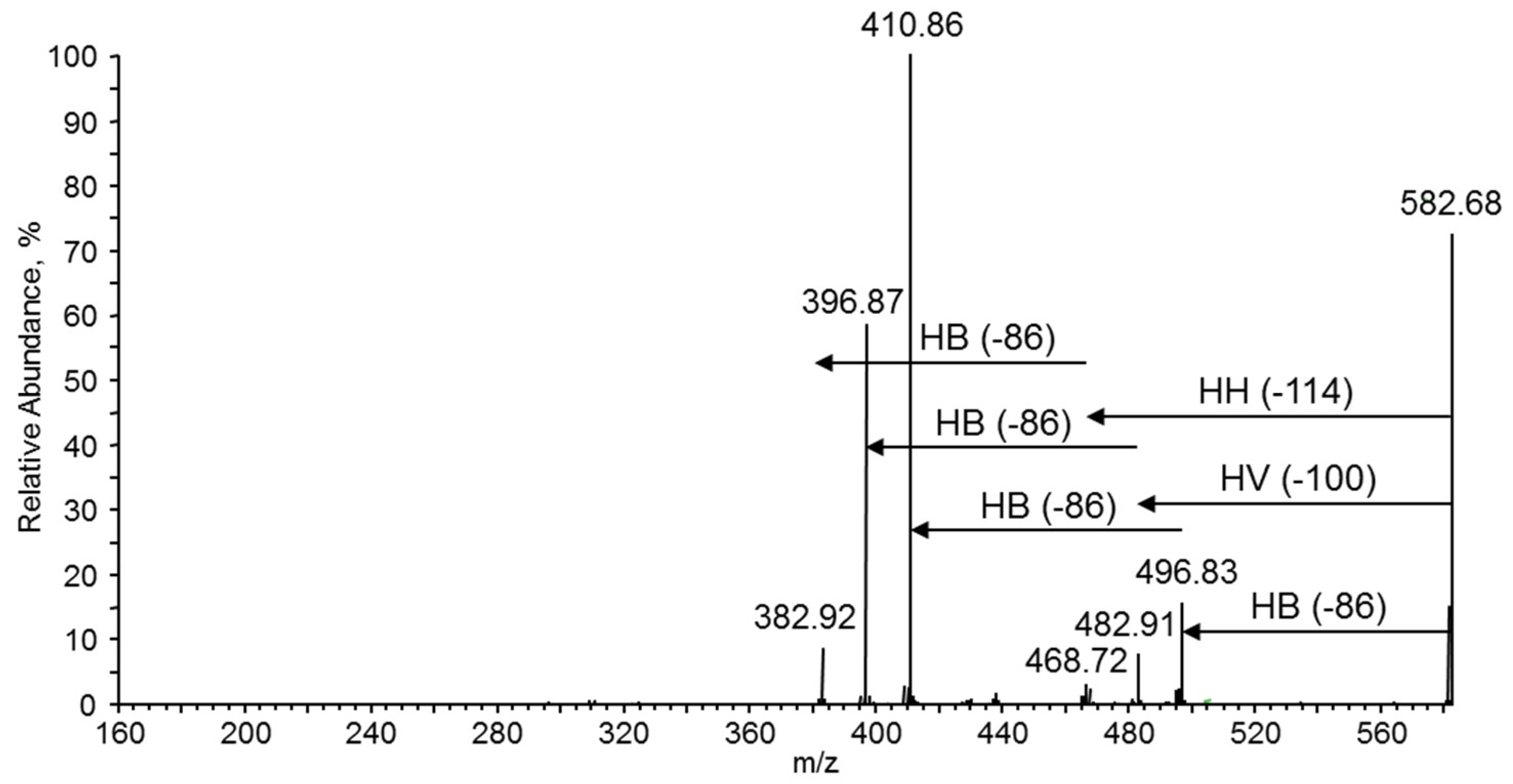
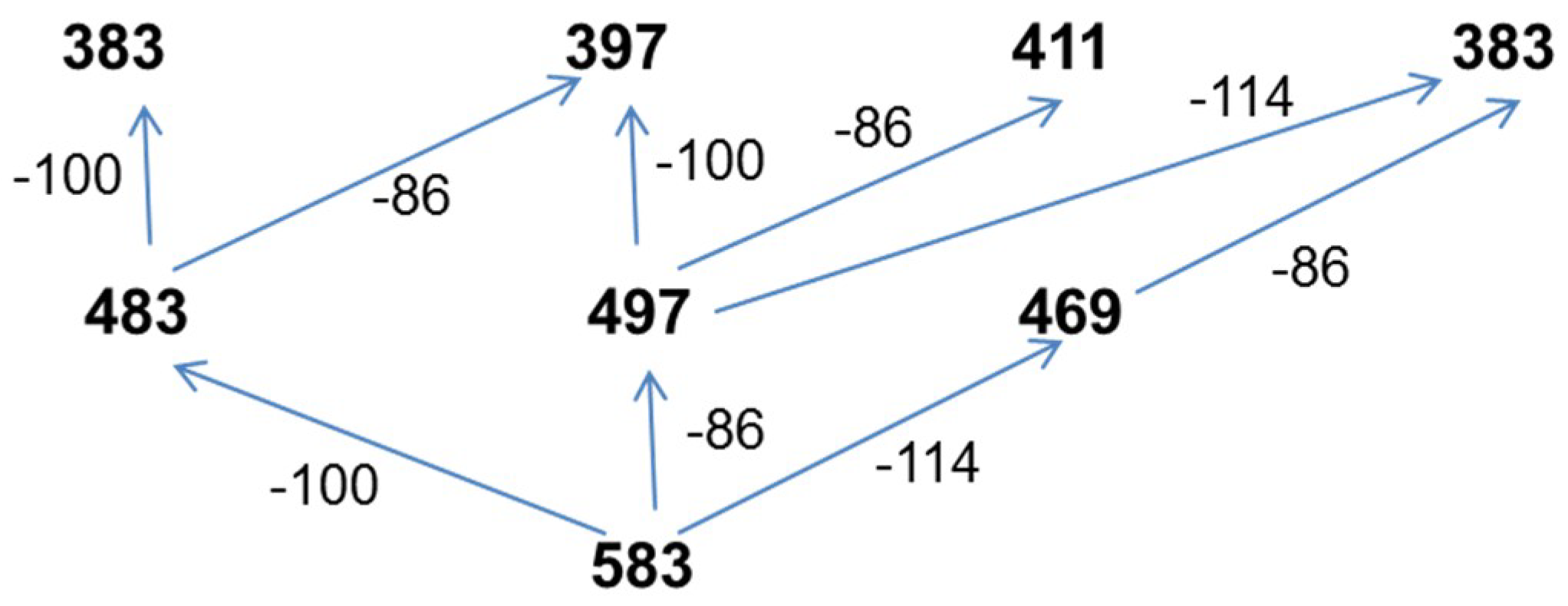
The ion at
m/z 583, presented in
Figure 10a, may corresponded to the isobaric potassium adduct ions containing two HV units or one HH unit [HB
5 HV
2 + K]
+ or [HB
5 HH +K]
+, respectively. Conversely, three fragmentation paths were observed in the case of the ESI-MS/MS spectrum of the parent ion at
m/z 583 (
Figure 12). Two series at
m/z 496, 411 and at
m/z 483, 383 were formed in the same way as described for fragmentation of the ion at
m/z 569 (
Figure 11). However, the third series at
m/z 467, 383 indicates that the first ion in this series is formed by the expulsion of 2-hexenoic acid (114 Da), which unequivocally confirms the presence of 3-hydroxyhexanoate (3-HH) units in the oligomer chain. Thus, the fragmentation experiment performed for the ion at
m/z 583 indicates the presence of three co-monomeric units in this oligomer; namely, 3-hydroxybutyrate and 3-hydroxyvalerate as well as 3-HH co-monomer units, which are randomly distributed along the oligomer chain. It is interesting to note that the fragmentation results are similar to those reported previously in the presence of oxidized polyethylene waxes as carbon sources for bacteria to produce PHAs, as described by Radecka et al. [
35].
The significance of the ratio of PHB to PHBV is that properties, such as melting point or mechanical strength, are changed; therefore, the applications of the bioplastic synthesized could be determined. In the case of PHAs, these applications include: General packaging of differing biodegradation rates, electrospinning materials, and 3D printing filaments [
7,
54]. PHAs that contain functional groups are also extremely appealing for biomedical usage as they allow for further chemical modification. This could be in the form of bioactive oligomers that distribute drugs using pH-controlled release [
55]. Polyethylene glycol (PEG) is a polyether that has been combined with PHA to alter its properties. Bulk polymerized PHBHHx/PEG poly (ether ester urethane) block copolymers have shown improved tissue compatibility, with greater physicochemical properties when used in vivo as biofilms [
56,
57]
. In addition, recombinant
E. coli enzymatic systems combined with fermentation optimization for PHA functionalization have been used to synthesize PEGylated PHB-co-PHHx copolymers with improved tissue regeneration abilities in comparison to PHB-co-PHHx and PHB, as well as “clickable” unsaturated PHA chains. Consequently, the PHA-polymer components for all these applications could originate from waste PS [
57,
58].
5. Conclusions
To assess the quality of the recycled GPPS and HIPS, an integrated approach was applied, involving different analytical strategies, such as GPC, FTIR, NMR, and ESI-MS/MS. From these analytical techniques, the chemical structure and morphological properties of the prodegraded PS0 and pretreated PS1–4 materials were noted. We have demonstrated that PHAs could be synthesized using PS0–3 samples as a carbon source. Even though there was a degree of mixing contamination of low molecular weight compounds from the PS samples used in the fermentation experiments of this study, to produce PHAs, waste prodegraded PS has proven to be a viable raw material. In fact, the contamination could alter the mechanical properties of the PHA synthesized, offering alternative applications.
We propose that the difference in samples may be evidence of the emission of volatiles during processing, and the formation of degradation products in other cases. The PS1 sample was the most energy efficient treated carbon source, with the most hydrophilic structure and produced the most biomass, with a promising 48% PHA yield. As such, unwanted PS could potentially join the group of carbon-rich industrial waste streams (“2nd generation feedstocks”) that can be used as an alternative to carbon sources that are derived from food and feed supplies. This method would not only reduce PHA production costs, and contribute towards safeguarding food and feed supplies in various disadvantaged parts of the world, but also help reduce the PS content in landfills. The aims of the EU, within the vision of a circular economy, are to reduce the leakage of plastics into the environment and recycle, which could be assisted by using oxo-degradation to form carbon sources for industrial fermentation. This proposed strategy could be employed alongside other sustainable chemical techniques to tackle sea-based marine litter particularly, including PS, GPPS, HIPS, and other types of plastic packaging.

 ,
, 















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}