Acrylated Chitosan Nanoparticles with Enhanced Mucoadhesion

Abstract
:
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Synthesis of Acrylated Chitosan
2.3. Synthesis of Fluorescein Isocyanate (FITC)-Labeled Polymers
2.4. Polymer Characterization
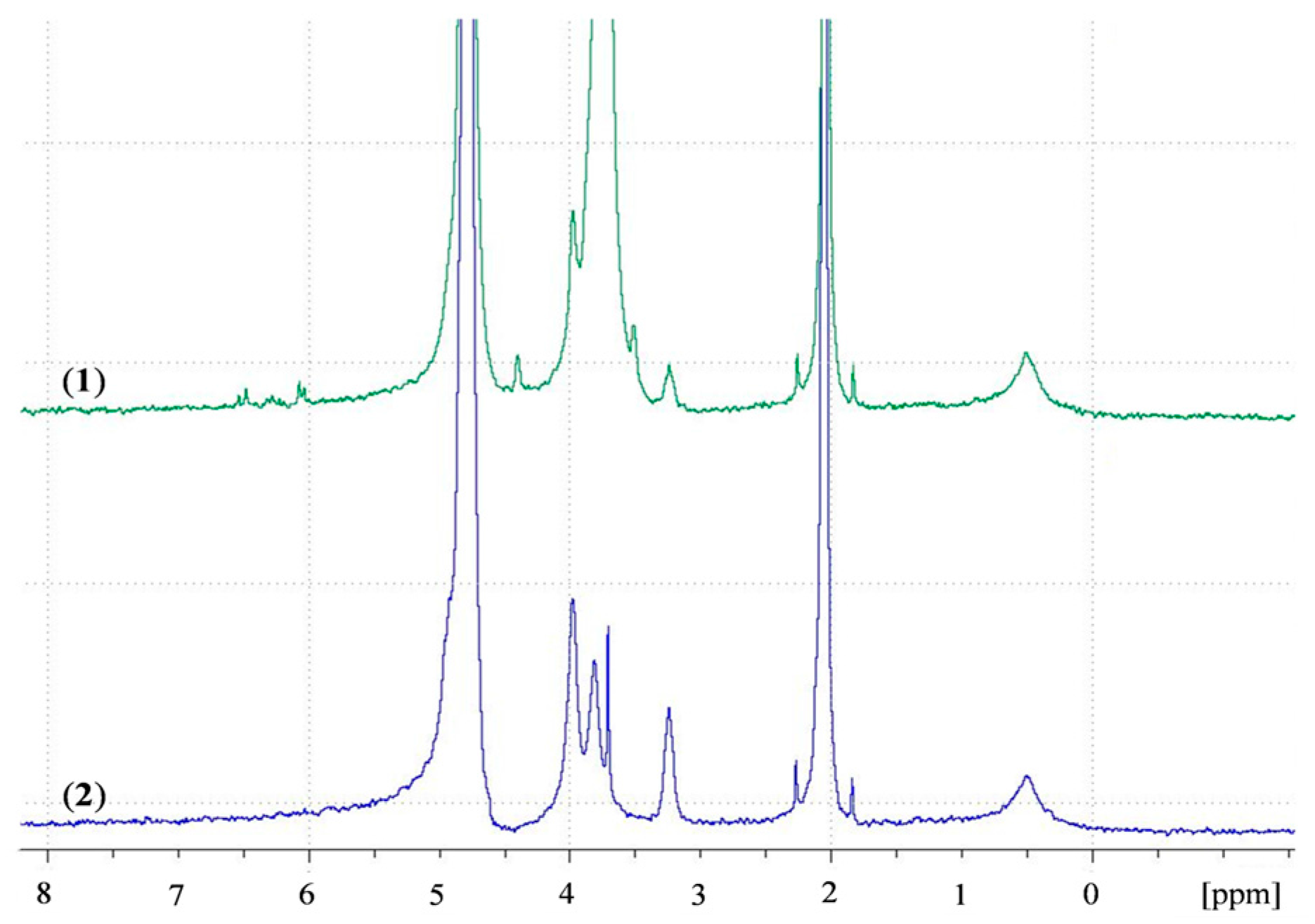
2.4.1. Nuclear Magnetic Resonance (NMR)
2.4.2. Fluorescamine Test for Quantitative Determination of Acrylate Groups
2.5. Fabrication of Nanoparticles
2.6. Nanoparticle Characterization
2.6.1. Size and Zeta Potential
2.6.2. Yield
2.7. Mucoadhesion Studies
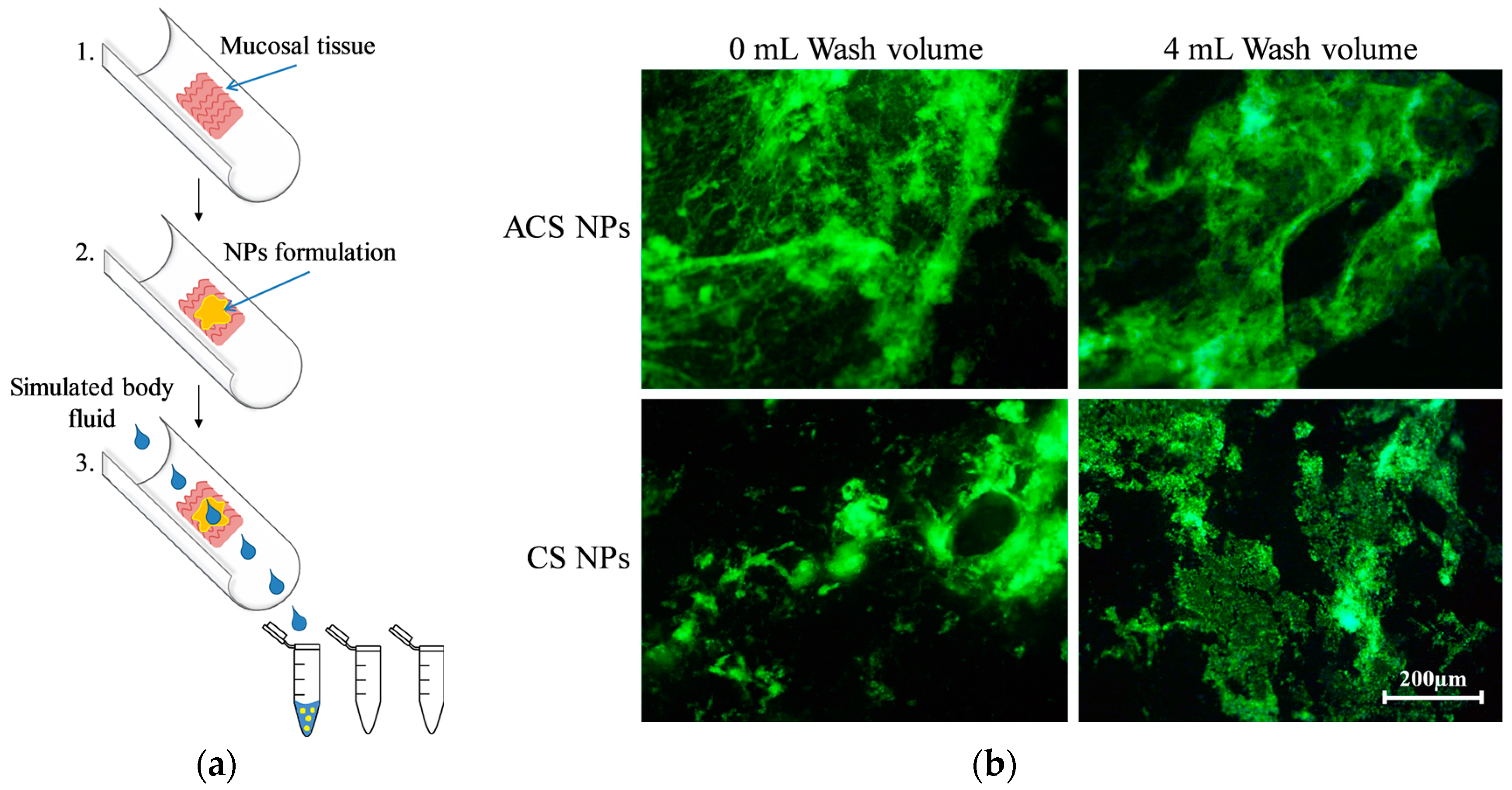
2.7.1. Nanoparticle Retention Studies
2.7.2. Nanoparticle-Mucin Aggregation
2.8. Statistical Data Analysis
3. Results and Discussion
3.1. Acrylation of Chitosan
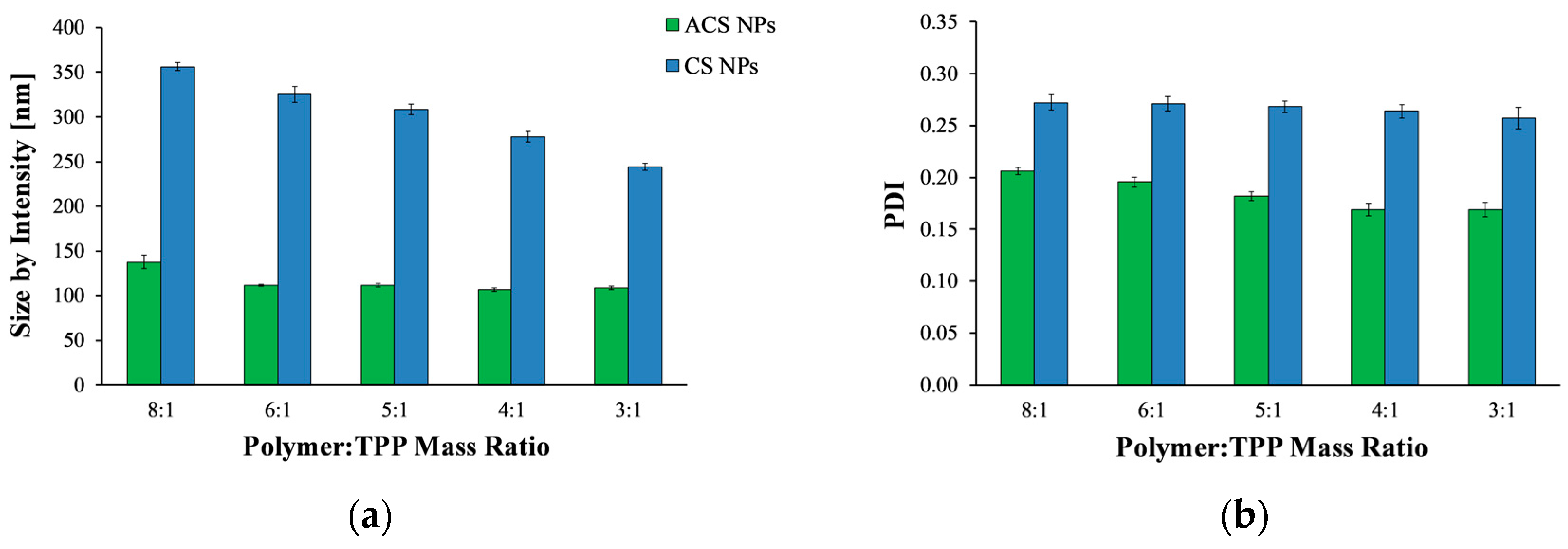
3.2. Fabrication of Nanoparticles
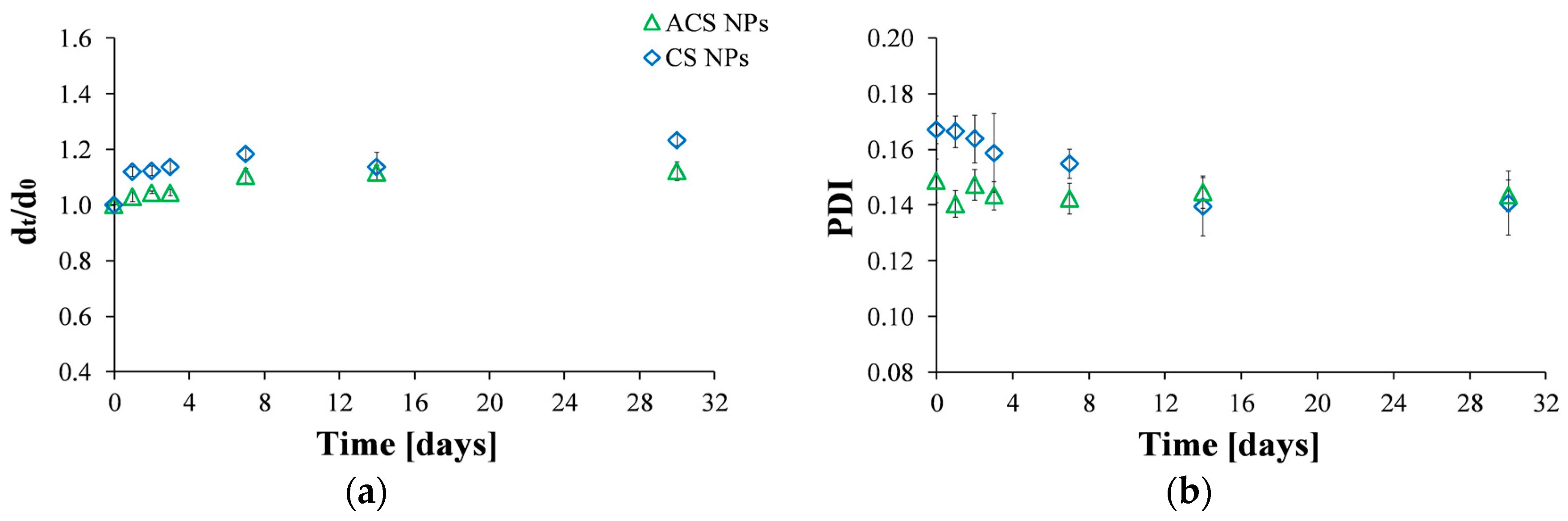
3.3. Stability of the Nanoparticles
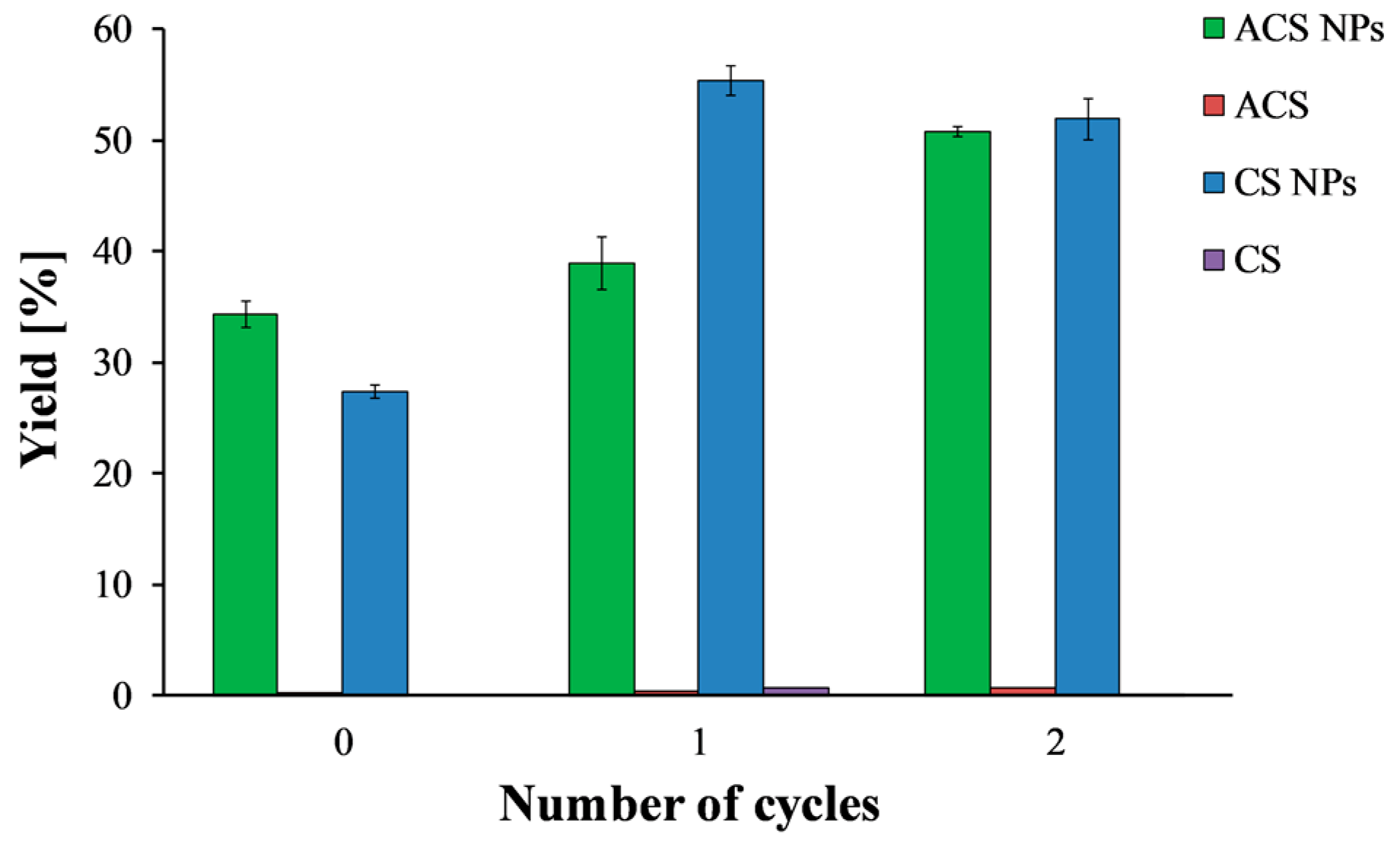
3.4. Nanoparticle Yield
3.5. Mucoadhesion Characterization
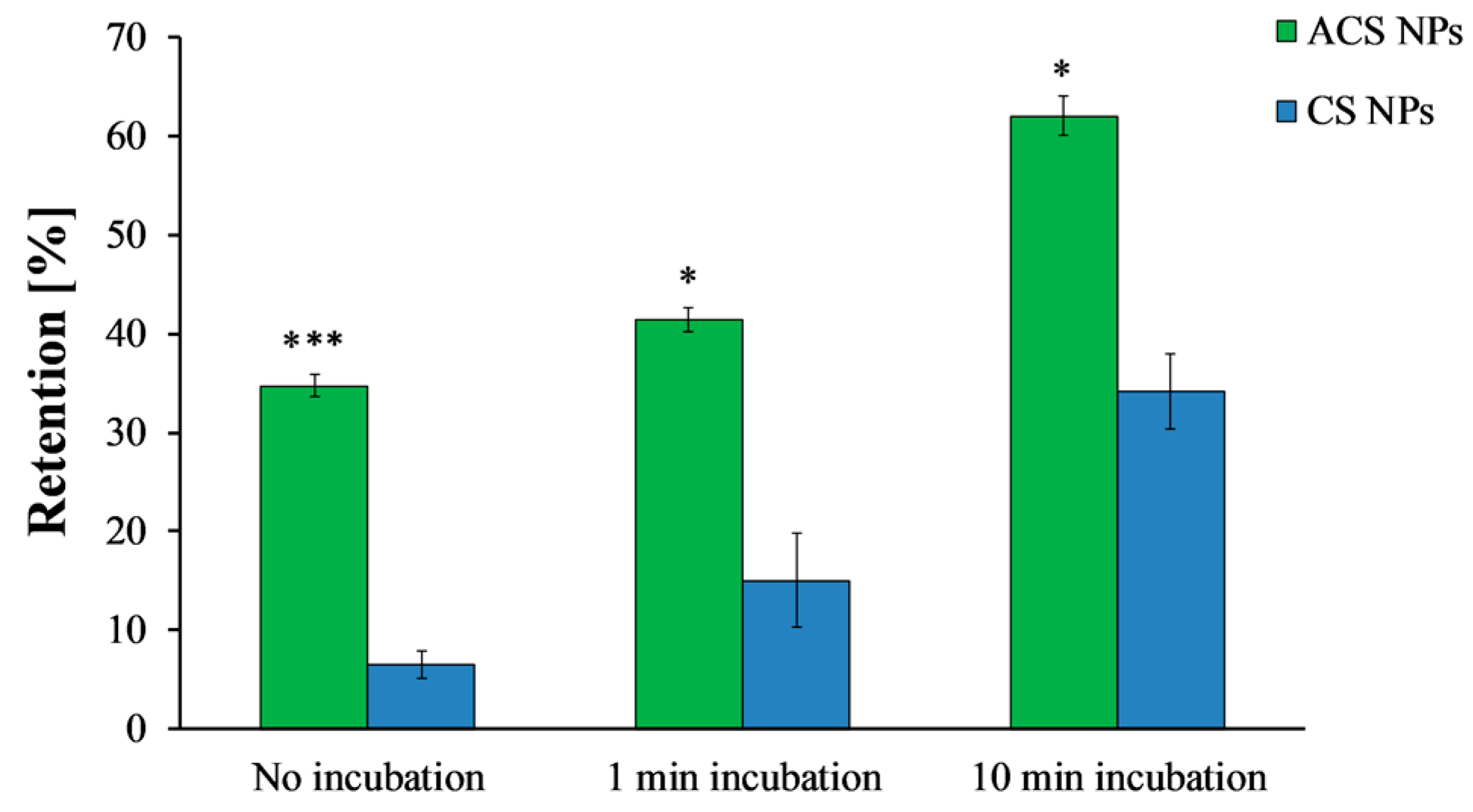
3.5.1. Nanoparticle Retention on Mucosal Surfaces
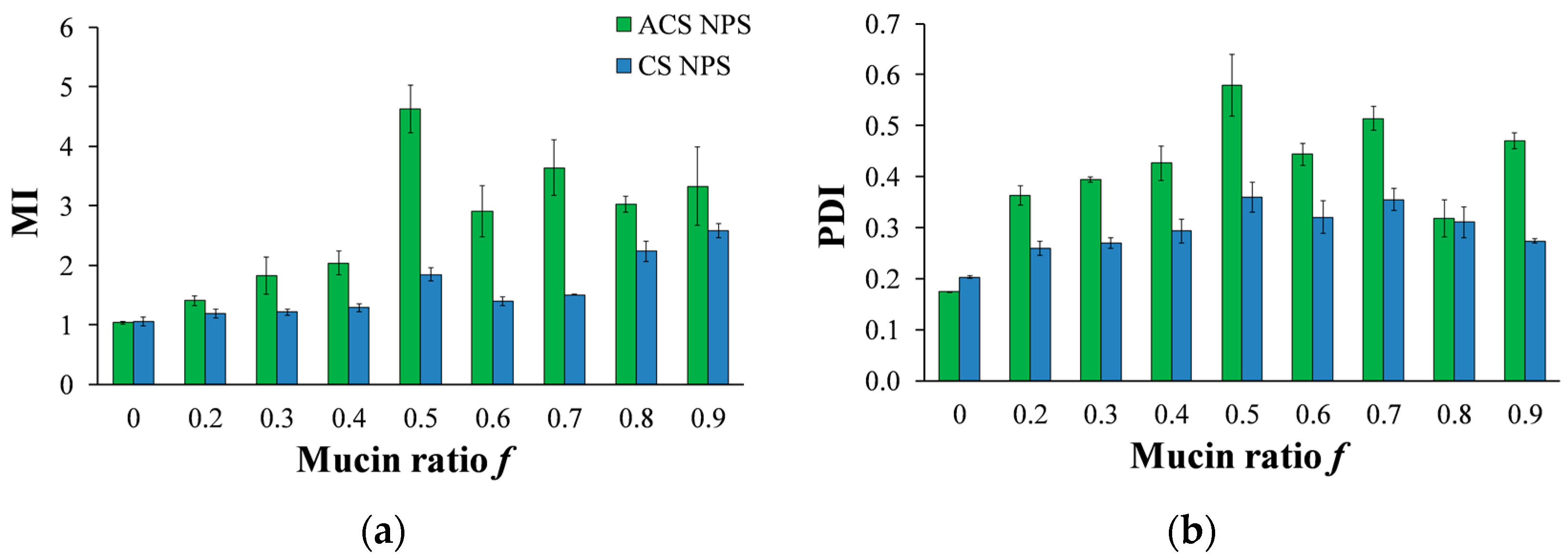
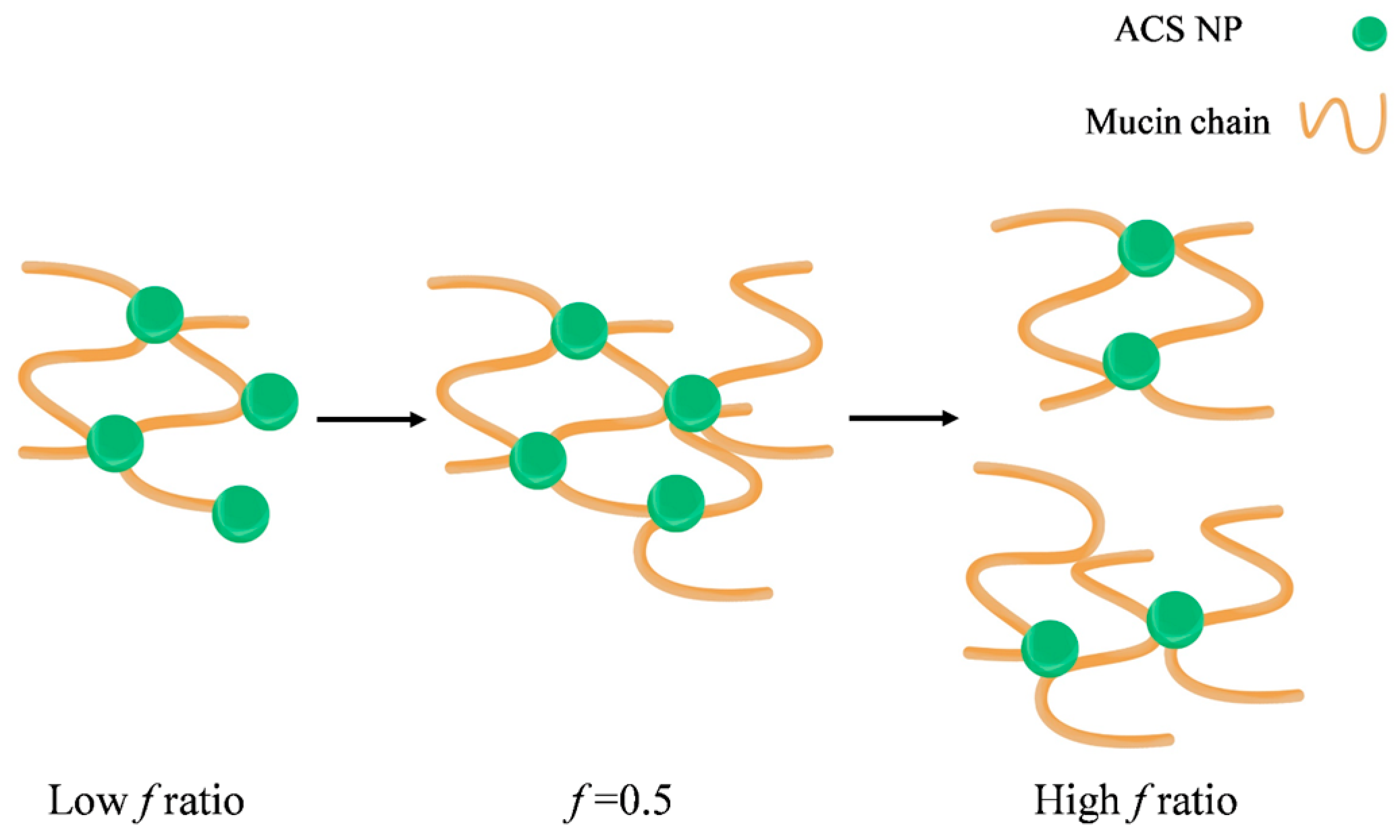
3.5.2. Nanoparticle-Mucin Aggregation
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Khutoryanskiy, V.V. Advances in Mucoadhesion and Mucoadhesive Polymers. Macromol. Biosci. 2011, 11, 748–764. [Google Scholar] [CrossRef] [PubMed]
- Das Neves, J.; Bahia, M.F.; Amiji, M.M.; Sarmento, B. Mucoadhesive nanomedicines: Characterization and modulation of mucoadhesion at the nanoscale. Expert Opin. Drug Deliv. 2011, 8, 1085–1104. [Google Scholar] [CrossRef] [PubMed]
- Andersen, T.; Mishchenko, E.; Flaten, G.E.; Sollid, J.U.E.; Mattsson, S.; Tho, I.; Škalko-Basnet, N. Chitosan-based nanomedicine to fight genital Candida Infections: Chitosomes. Mar. Drugs 2017, 15, 64. [Google Scholar] [CrossRef] [PubMed]
- Yeh, T.-H.; Hsu, L.-W.; Tseng, M.T.; Lee, P.-L.; Sonjae, K.; Ho, Y.-C.; Sung, H.-W. Mechanism and consequence of chitosan-mediated reversible epithelial tight junction opening. Biomaterials 2011, 32, 6164–6173. [Google Scholar] [CrossRef] [PubMed]
- Sogias, I.A.; Williams, A.C.; Khutoryanskiy, V.V. Why is chitosan mucoadhesive? Biomacromolecules 2008, 9, 1837–1842. [Google Scholar] [CrossRef] [PubMed]
- Maciel, V.B.V.; Yoshida, C.M.P.; Pereira, S.M.S.S.; Goycoolea, F.M.; Franco, T.T. Electrostatic Self-Assembled Chitosan-Pectin Nano-and Microparticles for Insulin Delivery. Molecules 2017, 22, 1707. [Google Scholar] [CrossRef] [PubMed]
- Jonassen, H.; Kjøniksen, A.L.; Hiorth, M. Effects of ionic strength on the size and compactness of chitosan nanoparticles. Colloid Polym. Sci. 2012, 290, 919–929. [Google Scholar] [CrossRef]
- Bernkop-Schnürch, A.; Weithaler, A.; Albrecht, K.; Greimel, A. Thiomers: Preparation and in vitro evaluation of a mucoadhesive nanoparticulate drug delivery system. Int. J. Pharm. 2006, 317, 76–81. [Google Scholar] [CrossRef] [PubMed]
- Chen, M.C.; Mi, F.L.; Liao, Z.X.; Hsiao, C.W.; Sonaje, K.; Chung, M.F.; Hsu, L.W.; Sung, H.W. Recent advances in chitosan-based nanoparticles for oral delivery of macromolecules. Adv. Drug Deliv. Rev. 2013, 65, 865–879. [Google Scholar] [CrossRef] [PubMed]
- Alamdarnejad, G.; Sharif, A.; Taranejoo, S.; Janmaleki, M.; Kalaee, M.R.; Dadgar, M.; Khakpour, M. Synthesis and characterization of thiolated carboxymethyl chitosan-graft-cyclodextrin nanoparticles as a drug delivery vehicle for albendazole. J. Mater. Sci. Mater. Med. 2013, 24, 1939–1949. [Google Scholar] [CrossRef] [PubMed]
- Dünnhaupt, S.; Barthelmes, J.; Köllner, S.; Sakloetsakun, D.; Shahnaz, G.; Düregger, A.; Bernkop-Schnürch, A. Thiolated nanocarriers for oral delivery of hydrophilic macromolecular drugs. Carbohydr. Polym. 2015, 117, 577–584. [Google Scholar] [CrossRef] [PubMed]
- Sunena; Mishra, D.; Singh, S.K.; Kumar, A. Formulation and optimization of mucoadhesive galantamine loaded nanoparticles. Pharm. Lett. 2016, 8, 206–212. [Google Scholar]
- Davidovich-Pinhas, M.; Bianco-Peled, H. Novel mucoadhesive system based on sulfhydryl-acrylate interactions. J. Mater. Sci. Mater. Med. 2010, 21, 2027–2034. [Google Scholar] [CrossRef] [PubMed]
- Davidovich-Pinhas, M.; Bianco-Peled, H. Alginate–PEGAc: A new mucoadhesive polymer. Acta Biomater. 2011, 7, 625–633. [Google Scholar] [CrossRef] [PubMed]
- Eshel-Green, T.; Bianco-Peled, H. Mucoadhesive acrylated block copolymers micelles for the delivery of hydrophobic drugs. Colloids Surf. B Biointerfaces 2016, 139, 42–51. [Google Scholar] [CrossRef] [PubMed]
- Shitrit, Y.; Bianco-Peled, H. Acrylated chitosan for mucoadhesive drug delivery systems. Int. J. Pharm. 2017, 517, 247–255. [Google Scholar] [CrossRef] [PubMed]
- Fwu-Long, M.I.; Shyu, S.S.; Lee, S.T.; Wong, T.B.I. Kinetic study of chitosan-tripolyphosphate complex reaction and acid-resistive properties of the chitosan-tripolyphosphate gel beads prepared by in-liquid curing method. J. Polym. Sci. Part B Polym. Phys. 1999, 37, 1551–1564. [Google Scholar] [CrossRef]
- Zhang, H.; Oh, M.; Allen, C.; Kumacheva, E. Monodisperse chitosan nanoparticles for mucosal drug delivery. Biomacromolecules 2004, 5, 2461–2468. [Google Scholar] [CrossRef] [PubMed]
- Dudhani, A.R.; Kosaraju, S.L. Bioadhesive chitosan nanoparticles: Preparation and characterization. Carbohydr. Polym. 2010, 81, 243–251. [Google Scholar] [CrossRef]
- Raskin, M.M.; Schlachet, I.; Sosnik, A. Mucoadhesive nanogels by ionotropic crosslinking of chitosan-g-oligo(NiPAam) polymeric micelles as novel drug nanocarriers. Nanomedicine (Lond.) 2016, 11, 217–233. [Google Scholar] [CrossRef] [PubMed]
- Tallury, P.; Kar, S.; Bamrungsap, S.; Huang, Y.-F.; Tan, W.; Santra, S. Ultra-small water-dispersible fluorescent chitosan nanoparticles: Synthesis, characterization and specific targeting. Chem. Commun. 2009, 2347–2349. [Google Scholar] [CrossRef] [PubMed]
- Udenfriend, S.; Stein, S.; Bohlen, P.; Dairman, W.; Leimgruber, W.; Weigele, M. Fluorescamine: A Reagent for Assay of Amino Acids, Peptides, Proteins, and Primary Amines in the Picomole Range. Science 1972, 178, 871–872. [Google Scholar] [CrossRef] [PubMed]
- Nasti, A.; Zaki, N.M.; De Leonardis, P.; Ungphaiboon, S.; Sansongsak, P.; Rimoli, M.G.; Tirelli, N. Chitosan/TPP and chitosan/TPP-hyaluronic acid nanoparticles: Systematic optimisation of the preparative process and preliminary biological evaluation. Pharm. Res. 2009, 26, 1918–1930. [Google Scholar] [CrossRef] [PubMed]
- Lim, S.; Seib, P.A. Preparation and Pasting Properties of Wheat and Corn Starch Phosphates. Cereal Chem. 1993, 70, 137–144. [Google Scholar] [CrossRef]
- Huang, Y.; Lapitsky, Y. Salt-assisted mechanistic analysis of chitosan/tripolyphosphate micro- and nanogel formation. Biomacromolecules 2012, 13, 3868–3876. [Google Scholar] [CrossRef] [PubMed]
- Antoniou, J.; Liu, F.; Majeed, H.; Qi, J.; Yokoyama, W.; Zhong, F. Physicochemical and morphological properties of size-controlled chitosan-tripolyphosphate nanoparticles. Colloids Surf. A Physicochem. Eng. Asp. 2015, 465, 137–146. [Google Scholar] [CrossRef]
- Eshel-Green, T.; Eliyahu, S.; Avidan-Shlomovich, S.; Bianco-Peled, H. PEGDA hydrogels as a replacement for animal tissues in mucoadhesion testing. Int. J. Pharm. 2016, 506, 25–34. [Google Scholar] [CrossRef] [PubMed]
- Jantratid, E.; Janssen, N.; Reppas, C.; Dressman, J.B. Dissolution media simulating conditions in the proximal human gastrointestinal tract: An update. Pharm. Res. 2008, 25, 1663–1676. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Gao, C. Preparation and properties of ionically cross-linked chitosan nanoparticles. Polym. Adv. Technol. 2009, 20, 613–619. [Google Scholar] [CrossRef]
- Shah, S.; Pal, A.; Kaushik, V.K.; Devi, S. Preparation and characterization of venlafaxine hydrochloride-loaded chitosan nanoparticles and in vitro release of drug. J. Appl. Polym. Sci. 2009, 112, 2876–2887. [Google Scholar] [CrossRef]
- Huang, Y.; Lapitsky, Y. Monovalent salt enhances colloidal stability during the formation of chitosan/tripolyphosphate microgels. Langmuir 2011, 27, 10392–10399. [Google Scholar] [CrossRef] [PubMed]
- Arzenšek, D.; Podgornik, R.; Kuzman, D. Dynamic light scattering and application to proteins in solutions. In Seminar; University of Ljubljana: Ljubljana, Slovenia, 2010; pp. 1–18. [Google Scholar]
- Masarudin, M.J.; Cutts, S.M.; Evison, B.J.; Phillips, D.R.; Pigram, P.J. Factors determining the stability, size distribution, and cellular accumulation of small, monodisperse chitosan nanoparticles as candidate vectors for anticancer drug delivery: Application to the passive encapsulation of [14C]-doxorubicin. Nanotechnol. Sci. Appl. 2015, 8, 67–80. [Google Scholar] [CrossRef] [PubMed]
- Riddick, T.M. Control of Colloid Stability through Zeta Potential; Zeta-Meter: Staunton, VA, USA, 1968. [Google Scholar]
- Hashad, R.A.; Ishak, R.A.H.; Fahmyb, S.; Mansour, S.; Geneidi, A.S. Chitosan-tripolyphosphate nanoparticles: Optimization of formulation parameters for improving process yield at a novel pH using artificial neural networks. Int. J. Biol. Macromol. 2016, 86, 50–58. [Google Scholar] [CrossRef] [PubMed]
- Rampino, A.; Borgogna, M.; Blasi, P.; Bellich, B.; Cesàro, A. Chitosan nanoparticles: Preparation, size evolution and stability. Int. J. Pharm. 2013, 455, 219–228. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, A.; Okada, R. The coagulation of particles in suspension by freezing-thawing—I. Effect of freezing-thawing conditions and other factors on coagulation. Colloid Polym. Sci. 1976, 254, 718–725. [Google Scholar] [CrossRef]
- Barb, W.G.; Mikucki, W. On the Coagulation of Polymer Latices by Freezing and Thawing. J. Polym. Sci. 1959, 37, 499–514. [Google Scholar] [CrossRef]
- Rao, K.V.; Buri, P. A novel in situ method to test polymers and coated microparticles for bioadhesion. Int. J. Pharm. 1989, 52, 265–270. [Google Scholar] [CrossRef]
- Albrecht, K.; Zirm, E.J.; Palmberger, T.F.; Schlocker, W.; Bernkop-Schnürch, A. Preparation of thiomer microparticles and in vitro evaluation of parameters influencing their mucoadhesive properties. Drug Dev. Ind. Pharm. 2006, 32, 1149–1157. [Google Scholar] [CrossRef] [PubMed]
- Storha, A.; Mun, E.A.; Khutoryanskiy, V. V Synthesis of thiolated and acrylated nanoparticles using thiol-ene click chemistry: Towards novel mucoadhesive materials for drug delivery. RSC Adv. 2013, 3, 12275–12279. [Google Scholar] [CrossRef]
- Shaikh, R.; Raj Singh, T.R.; Garland, M.J.; Woolfson, A.D.; Donnelly, R.F. Mucoadhesive drug delivery systems. J. Pharm. Bioallied Sci. 2011, 3, 89–100. [Google Scholar] [CrossRef] [PubMed]
- Dust, J.M.; Fang, Z.H.; Harris, J.M. Proton NMR Characterization of Poly(ethylene Glycols) and Derivatives. Macromolecules 1990, 23, 3742–3746. [Google Scholar] [CrossRef]
- Ma, G.; Zhang, X.; Han, J.; Song, G.; Nie, J. Photo-polymeriable chitosan derivative prepared by Michael reaction of chitosan and polyethylene glycol diacrylate (PEGDA). Int. J. Biol. Macromol. 2009, 45, 499–503. [Google Scholar] [CrossRef] [PubMed]
- Teng, D.-Y.; Wu, Z.-M.; Zhang, X.-G.; Wang, Y.-X.; Zheng, C.; Wang, Z.; Li, C.-X. Synthesis and characterization of in situ cross-linked hydrogel based on self-assembly of thiol-modified chitosan with PEG diacrylate using Michael type addition. Polymer 2010, 51, 639–646. [Google Scholar] [CrossRef]
- Menchicchi, B.; Fuenzalida, J.P.; Bobbili, K.B.; Hensel, A.; Swamy, M.J.; Goycoolea, F.M. Structure of Chitosan determines its interactions with mucin. Biomacromolecules 2014, 15, 3550–3558. [Google Scholar] [CrossRef] [PubMed]
- Bansil, R.; Turner, B.S. Mucin structure, aggregation, physiological functions and biomedical applications. Curr. Opin. Colloid Interface Sci. 2006, 11, 164–170. [Google Scholar] [CrossRef]
- Rossi, S.; Ferrari, F.; Bonferoni, M.C.; Caramella, C. Characterization of chitosan hydrochloride–mucin rheological interaction: Influence of polymer concentration and polymer: Mucin weight ratio. Eur. J. Pharm. Sci. 2001, 12, 479–485. [Google Scholar] [CrossRef]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| CS Concentration (mg/mL) | TPP Concentration (mg/mL) | Size by Intensity (nm) |
|---|---|---|
| 2 | 1 | 354.6 ± 4.5 |
| 2 | 0.5 | 317.0 ± 6.6 |
| 2 | 0.25 | 249.0 ± 3.4 |
| 1 | 0.5 | 228.6 ± 0.8 |
| 1 | 0.25 | 209.4 ± 3.0 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Eliyahu, S.; Aharon, A.; Bianco-Peled, H. Acrylated Chitosan Nanoparticles with Enhanced Mucoadhesion. Polymers 2018, 10, 106. https://doi.org/10.3390/polym10020106
Eliyahu S, Aharon A, Bianco-Peled H. Acrylated Chitosan Nanoparticles with Enhanced Mucoadhesion. Polymers. 2018; 10(2):106. https://doi.org/10.3390/polym10020106
Chicago/Turabian StyleEliyahu, Shaked, Anat Aharon, and Havazelet Bianco-Peled. 2018. "Acrylated Chitosan Nanoparticles with Enhanced Mucoadhesion" Polymers 10, no. 2: 106. https://doi.org/10.3390/polym10020106