Synthesis of Polystyrene-Coated Superparamagnetic and Ferromagnetic Cobalt Nanoparticles



Abstract
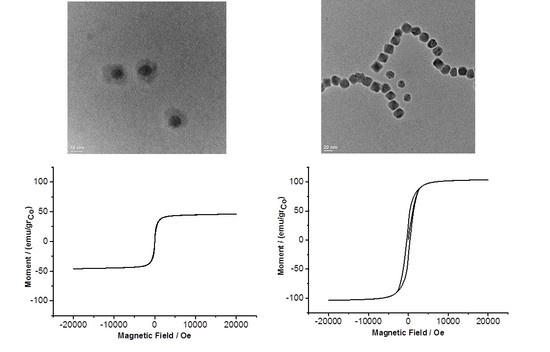
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
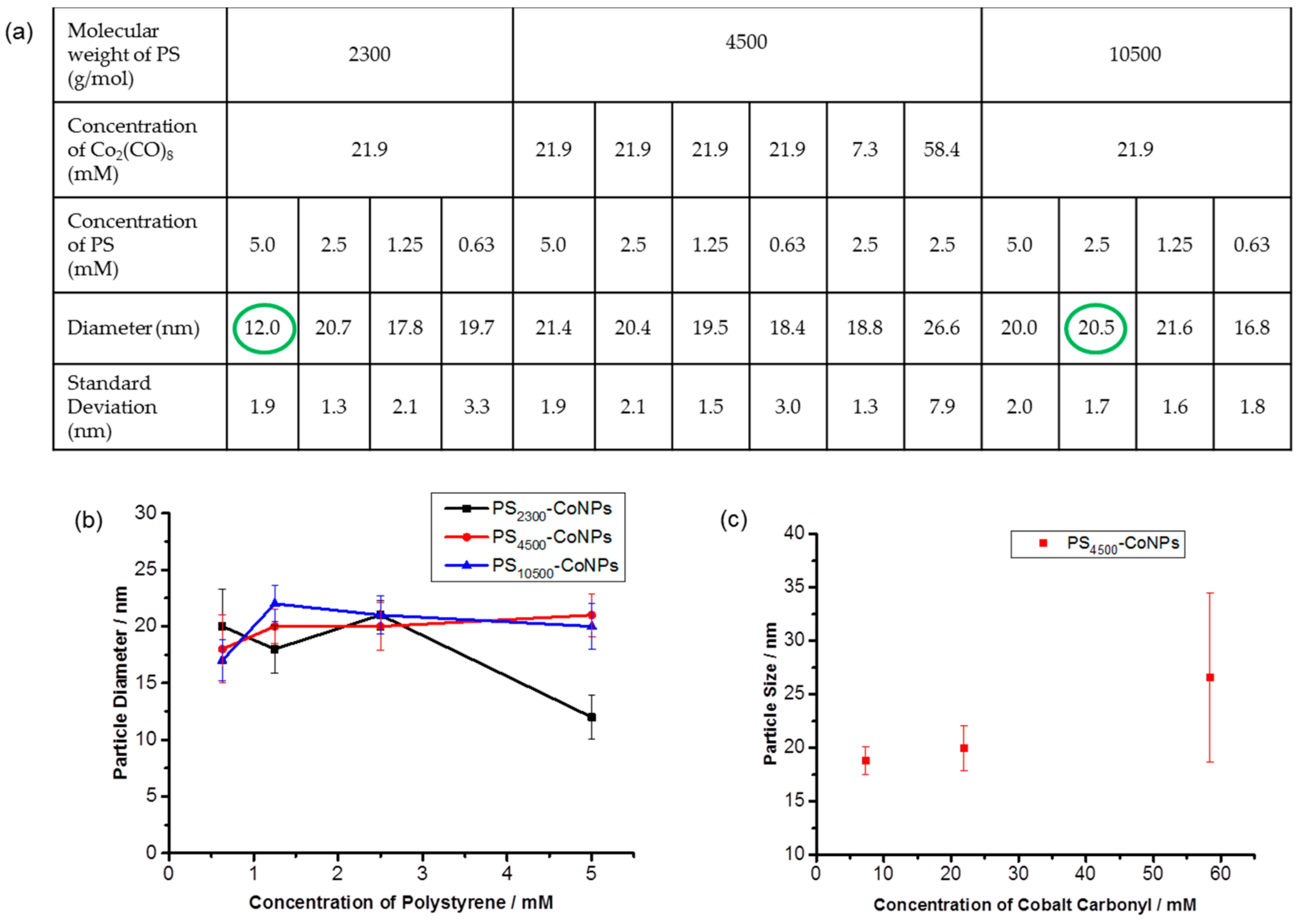{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Synthesis of Amine End-Functionalized Polystyrene (PS-NH2)
2.2.1. Synthesis of 4-(Chloromethyl)benzyl Phthalimide 2
2.2.2. Synthesis of Benzyl Phthalimide End-Functionalized Polystyrene 3
2.2.3. Synthesis of Amine End-Functionalized Polystyrene 4 (PS-NH2)
2.3. Synthesis of PS-Co NPs with Amine End-Functionalized Polystyrene as Surfactant
2.4. Instruments
3. Results and Discussion
3.1. Synthesis and Characterization of PS-NH2
3.2. Synthesis and Characterization of PS–Co NPs
3.3. Magnetic Properties of PS-Co NPs
3.4. Wide Angle X-ray Scattering of PS-Co NPs
3.5. Calculation of Surface Polymer Density
3.6. Stability Study
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- De Dios, A.S.; Díaz-García, M.E. Multifunctional nanoparticles: Analytical prospects. Anal. Chim. Acta 2010, 666, 1–22. [Google Scholar] [CrossRef] [PubMed]
- Frey, N.A.; Peng, S.; Cheng, K.; Sun, S. Magnetic nanoparticles: synthesis, functionalization, and applications in bioimaging and magnetic energy storage. Chem. Soc. Rev. 2009, 38, 2532–2542. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Corot, C.; Robert, P.; Idée, J.M.; Port, M. Recent advances in iron oxide nanocrystal technology for medical imaging. Adv. Drug Deliv. Rev. 2006, 58, 1471–1504. [Google Scholar] [CrossRef] [PubMed]
- Mornet, S.; Vasseur, S.; Grasset, F.; Duguet, E. Magnetic nanoparticle design for medical diagnosis and therapy. J. Mater. Chem. 2004, 14, 2161–2175. [Google Scholar] [CrossRef]
- Bao, Y.; An, W.; Heath Turner, C.; Krishnan, K.M. The critical role of surfactants in the growth of cobalt nanoparticles. Langmuir 2010, 26, 478–483. [Google Scholar] [CrossRef] [PubMed]
- Bao, Y.; Beerman, M.; Pakhomov, A.B.; Krishnan, K.M. Controlled Crystalline Structure and Surface Stability of Cobalt Nanocrystals Controlled Crystalline Structure and Surface Stability of Cobalt Nanocrystals. J. Phys. Chem. 2005, 109, 7220–7222. [Google Scholar] [CrossRef] [PubMed]
- Sun, S.; Murray, C.B. Synthesis of monodisperse cobalt nanocrystals and their assembly into magnetic superlattices (invited). J. Appl. Phys. 1999, 85, 4325–4330. [Google Scholar] [CrossRef]
- Tan, L.; Liu, B.; Siemensmeyer, K.; Glebe, U.; Böker, A. Synthesis of thermo-responsive nanocomposites of superparamagnetic cobalt nanoparticles/poly(N-isopropylacrylamide). J. Colloid Interface Sci. 2018, 526, 124–134. [Google Scholar] [CrossRef] [PubMed]
- Puntes, V.F.; Zanchet, D.; Erdonmez, C.K.; Alivisatos, A.P. Synthesis of hcp-Co nanodisks. J. Am. Chem. Soc. 2002, 124, 12874–12880. [Google Scholar] [CrossRef] [PubMed]
- Iablokov, V.; Beaumont, S.K.; Alayoglu, S.; Pushkarev, V.V.; Specht, C.; Gao, J.; Alivisatos, A.P.; Kruse, N.; Somorjai, G. A Size-controlled model Co nanoparticle catalysts for CO2 hydrogenation: Synthesis, characterization, and catalytic reactions. Nano Lett. 2012, 12, 3091–3096. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pinna, N.; Hochepied, J.F.; Niederberger, M.; Gregg, M. Chemistry and physics of metal oxide nanostructures. Phys. Chem. Chem. Phys. 2009, 11, 3607. [Google Scholar] [CrossRef] [PubMed]
- Mourdikoudis, S.; Liz-Marzán, L.M. Oleylamine in nanoparticle synthesis. Chem. Mater. 2013, 25, 1465–1476. [Google Scholar] [CrossRef]
- Puntes, V.F.; Krishanan, K.M.; Alivisatos, A.P. Colloidal Nanocrystal shape and size control. Science 2001, 291, 2115–2117. [Google Scholar] [CrossRef] [PubMed]
- Cheng, G.; Puntes, V.F.; Guo, T. Synthesis and self-assembled ring structures of Ni nanocrystals. J. Colloid Interface Sci. 2006, 293, 430–436. [Google Scholar] [CrossRef] [PubMed]
- Thomas, J.R. Preparation and magnetic properties of colloidal cobalt particles. J. Appl. Phys. 1966, 37, 2914–2915. [Google Scholar] [CrossRef]
- Diana, F.S.; Lee, S.H.; Petroff, P.M.; Kramer, E.J. Fabrication of hcp-Co nanocrystals via rapid pyrolysis in inverse PS-b-PVP micelles and thermal annealing. Nano Lett. 2003, 3, 891–895. [Google Scholar] [CrossRef]
- Valetsky, P.M.; Yanovskaya, I.M.; Obolonkova, E.S. Cobalt nanoparticles in block copolymer micelles: Preparation and properties. Polymer 1996, 2, 1–6. [Google Scholar] [CrossRef]
- Burke, N.A.D.; Stöver, H.D.H.; Dawson, F.P. Magnetic nanocomposites: Preparation and characterization of polymer-coated iron nanoparticles. Chem. Mater. 2002, 14, 4752–4761. [Google Scholar] [CrossRef]
- Liu, G.; Yan, X.; Lu, Z.; Curda, S.A.; Lal, J. One-pot synthesis of block copolymer coated cobalt nanocrystals. Chem. Mater. 2005, 17, 4985–4991. [Google Scholar] [CrossRef]
- Griffiths, C.H.; O’Horo, M.P.; Smith, T.W. The structure, magnetic characterization, and oxidation of colloidal iron dispersions. J. Appl. Phys. 1979, 50, 7108–7115. [Google Scholar] [CrossRef]
- Hess, P.H.; Parker, P.H. Polymers for stabilization of colloidal cobalt particles. J. Appl. Polym. Sci. 1966, 10, 1915–1927. [Google Scholar] [CrossRef]
- Ostermann, J.; Schmidtke, C.; Wolter, C.; Merkl, J.P.; Kloust, H.; Weller, H. Tailoring the ligand shell for the control of cellular uptake and optical properties of nanocrystals. Beilstein J. Nanotechnol. 2015, 6, 232–242. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tromsdorf, U.I.; Bruns, O.T.; Salmen, S.C.; Beisiegel, U.; Weller, H. A Highly Effective, Nontoxic T1 MR Contrast Agent Based on Ultrasmall PEGylated Iron Oxide Nanoparticles. Nano Lett. 2009, 9, 4434–4440. [Google Scholar] [CrossRef] [PubMed]
- Dobson, J. Magnetic micro- and nano-particle-based targeting for drug and gene delivery. Nanomedicine 2006, 1, 31–37. [Google Scholar] [CrossRef] [PubMed]
- Mornet, S.; Vasseur, S.; Grasset, F.; Veverka, P.; Goglio, G.; Demourgues, A.; Portier, J.; Pollert, E.; Duguet, E. Magnetic nanoparticle design for medical applications. Prog. Solid State Chem. 2006, 34, 237–247. [Google Scholar] [CrossRef]
- Pyun, J. Nanocomposite materials from functional polymers and magnetic colloids. Polym. Rev. 2007, 47, 231–263. [Google Scholar] [CrossRef]
- Korth, B.D.; Keng, P.Y.; Shim, I.; Tang, C.; Kowalewski, T.; Pyun, J. Synthesis, assembly, and functionalization of polymer-coated ferromagnetic nanoparticles. ACS Symp. Ser. 2008, 996, 272–285. [Google Scholar] [CrossRef]
- Zalich, M.A.; Vadala, M.L.; Riffle, J.S.; Saunders, M.; St. Pierre, T.G. Structural and magnetic properties of cobalt nanoparticles encased in siliceous shells. Chem. Mater. 2007, 19, 6597–6604. [Google Scholar] [CrossRef]
- Safran, S.A. Ferrofluids: Magnetic strings and networks. Nat. Mater. 2003, 2, 71–72. [Google Scholar] [CrossRef] [PubMed]
- Kolhatkar, A.G.; Jamison, A.C.; Litvinov, D.; Willson, R.C.; Lee, T.R. Tuning the magnetic properties of nanoparticles. Int. J. Mol. Sci. 2013, 14, 15977–16009. [Google Scholar] [CrossRef] [PubMed]
- Korth, B.D.; Keng, P.; Shim, I.; Bowles, S.E.; Tang, C.; Kowalewski, T.; Nebesny, K.W.; Pyun, J. Polymer-coated ferromagnetic colloids from well-defined macromolecular surfactants and assembly into nanoparticle chains. J. Am. Chem. Soc. 2006, 128, 6562–6563. [Google Scholar] [CrossRef] [PubMed]
- Keng, P.Y.; Shim, I.; Korth, B.D.; Douglas, J.F.; Pyun, J. Synthesis and self-assembly of polymer-coated ferromagnetic nanoparticles. ACS Nano 2007, 1, 279–292. [Google Scholar] [CrossRef] [PubMed]
- Bull, M.M.; Chung, W.J.; Anderson, S.R.; Kim, S.; Shim, I.-B.; Paik, H.; Pyun, J. Synthesis of ferromagnetic polymer coated nanoparticles on multi-gram scale with tunable particle size. J. Mater. Chem. 2010, 20, 6023. [Google Scholar] [CrossRef]
- di Lena, F.; Matyjaszewski, K. Transition metal catalysts for controlled radical polymerization. Prog. Polym. Sci. 2010, 35, 959–1021. [Google Scholar] [CrossRef]
- Braunecker, W.A.; Matyjaszewski, K. Controlled/living radical polymerization: Features, developments, and perspectives. Prog. Polym. Sci. 2007, 32, 93–146. [Google Scholar] [CrossRef]
- Wang, J.S.; Matyjaszewski, K. Controlled/“living” radical polymerization. Atom transfer radical polymerization in the presence of transition-metal complexes. J. Am. Chem.Soc. 1995, 117, 5614–5615. [Google Scholar] [CrossRef]
- Matyjaszewski, K. Bulk Atom Transfer Radical Polymerization. Chem. Rev. 2001, 101, 96–112. [Google Scholar] [CrossRef]
- Wu, L.; Glebe, U.; Böker, A. Surface-initiated controlled radical polymerizations from silica nanoparticles, gold nanocrystals, and bionanoparticles. Polym. Chem. 2015, 6, 5143–5184. [Google Scholar] [CrossRef]
- Matyjaszewski, K.; Tsarevsky, N.V. Nanostructured functional materials prepared by atom transfer radical polymerization. Nat. Chem. 2009, 1, 276–288. [Google Scholar] [CrossRef] [PubMed]
- Coessens, V.; Pintauer, T.; Matyjaszewski, K. Functional polymers by atom transfer radical polymerization. Prog. Polym. Sci. 2001, 26, 337–377. [Google Scholar] [CrossRef]
- Nigh, W.G. The Gabriel Synthesis of Benzylamine. J. Chem. Educ. 1975, 50, 670–671. [Google Scholar] [CrossRef]
- Shevchenko, E.V.; Talapin, D.V.; Schnablegger, H.; Kornowski, A.; Festin, Ö.; Svedlindh, P.; Haase, M.; Weller, H. Study of nucleation and growth in the organometallic synthesis of magnetic alloy nanocrystals: The role of nucleation rate in size control of CoPt3 nanocrystals. J. Am. Chem. Soc. 2003, 125, 9090–9101. [Google Scholar] [CrossRef] [PubMed]
- Hyeon, T.; Su, S.L.; Park, J.; Chung, Y.; Hyon, B.N. Synthesis of highly crystalline and monodisperse maghemite nanocrystallites without a size-selection process. J. Am. Chem. Soc. 2001, 123, 12798–12801. [Google Scholar] [CrossRef] [PubMed]
- Leff, D.V.; Ohara, P.C.; Heath, J.R.; Gelbart, W.M. Thermodynamic Control of Gold Nanocrystal Size: Experiment and Theory. J. Phys. Chem. 1995, 99, 7036–7041. [Google Scholar] [CrossRef]
- Talapin, D.V.; Rogach, A.L.; Haase, M.; Weller, H. Evolution of an ensemble of nanoparticles in a colloidal solution: Theoretical study. J. Phys. Chem. B 2001, 105, 12278–12285. [Google Scholar] [CrossRef]
- Peng, Z.A.; Peng, X. Mechanisms of the shape evolution of CdSe nanocrystals. J. Am. Chem. Soc. 2001, 123, 1389–1395. [Google Scholar] [CrossRef]
- Peng, X.; Wickham, J.; Alivisatos, A.P. Kinetics of II-VI and III-V colloidal semiconductor nanocrystal growth: “Focusing” of size distributions. J. Am. Chem. Soc. 1998, 120, 5343–5344. [Google Scholar] [CrossRef]
- Thanh, N.T.K.; Maclean, N.; Mahiddine, S. Mechanisms of nucleation and growth of nanoparticles in solution. Chem. Rev. 2014, 114, 7610–7630. [Google Scholar] [CrossRef] [PubMed]
- Talapin, D.V.; Mekis, I.; Götzinger, S.; Kornowski, A.; Benson, O.; Weller, H. CdSe/CdS/ZnS and CdSe/ZnSe/ZnS core-shell-shell nanocrystals. J. Phys. Chem. B 2004, 108, 18826–18831. [Google Scholar] [CrossRef]
- Alagiri, M.; Muthamizhchelvan, C.; Hamid, S.B.A. Synthesis of superparamagnetic cobalt nanoparticles through solvothermal process. J. Mater. Sci. Mater. Electron 2013, 24, 4157–4160. [Google Scholar] [CrossRef]
- Srikala, D.; Singh, V.N.; Banerjee, A.; Mehta, B.R.; Patnaik, S. Control of magnetism in cobalt nanoparticles by oxygen passivation. J. Phys. Chem. C 2008, 112, 13882–13885. [Google Scholar] [CrossRef]
- Tracy, J.B.; Weiss, D.N.; Dinega, D.P.; Bawendi, M.G. Exchange biasing and magnetic properties of partially and fully oxidized colloidal cobalt nanoparticles. Phys. Rev. B - Condens. Matter Mater. Phys. 2005, 72, 1–8. [Google Scholar] [CrossRef]
- Childress, J.R.; Chien, C.L. Reentrant magnetic behavior in fcc Co-Cu alloys. Phys. Rev. B 1991, 43, 8089–8093. [Google Scholar] [CrossRef]
- Shafi, K.V.P.M.; Gedanken, A.; Prozorov, R.; Balogh, J. Sonochemical Preparation and Size-Dependent Properties of Nanostructured CoFe2O4 Particles. Chem. Mater. 1998, 10, 3445–3450. [Google Scholar] [CrossRef]
- Van Leeuwen, D.A.; Van Ruitenbeek, J.M.; De Jongh, L.J.; Ceriotti, A.; Pacchioni, G.; Häberlen, O.D.; Rösch, N. Quenching of magnetic moments by ligand-metal interactions in nanosized magnetic metal clusters. Phys. Rev. Lett. 1994, 73, 1432–1435. [Google Scholar] [CrossRef] [PubMed]
- Hwang, J.H.; Dravid, V.P.; Teng, M.H.; Host, J.J.; Elliott, B.R.; Johnson, D.L.; Mason, T.O. Magnetic properties of graphitically encapsulated nickel nanocrystals. J. Mater. Res. 1997, 12, 1076–1082. [Google Scholar] [CrossRef]
- Mørup, S. Superparamagnetism and spin glass ordering in magnetic nanocomposites. EPL 1994, 28, 671–676. [Google Scholar] [CrossRef]
- Chen, D.H.; Wu, S.H. Synthesis of nickel nanoparticles in water-in-oil microemulsions. Chem. Mater. 2000, 12, 1354–1360. [Google Scholar] [CrossRef]
- Shamim, N.; Hong, L.; Hidajat, K.; Uddin, M.S. Thermosensitive polymer (N-isopropylacrylamide) coated nanomagnetic particles: Preparation and characterization. Colloids Surf. B: Biointerfaces 2007, 55, 51–58. [Google Scholar] [CrossRef] [PubMed]
- Dinega, D.P.; Bawendi, M.G. A solution-phase chemical approach to a new crystal structure of cobalt. Angew. Chem. Int. Ed. 1999, 38, 1788–1791. [Google Scholar] [CrossRef]
- Park, J.I.; Kang, N.J.; Jun, Y.W.; Oh, S.J.; Ri, H.C.; Cheon, J. Superlattice and magnetism directed by the size and shape of nanocrystals. ChemPhysChem 2002, 3, 543–547. [Google Scholar] [CrossRef]
- Mansfield, E.; Tyner, K.M.; Poling, C.M.; Blacklock, J.L. Determination of nanoparticle surface coatings and nanoparticle purity using microscale thermogravimetric analysis. Anal. Chem. 2014, 86, 1478–1484. [Google Scholar] [CrossRef] [PubMed]
- Prakash, A.; Zhu, H.; Jones, C.J.; Benoit, D.N.; Ellsworth, A.Z.; Bryant, E.L.; Colvin, V.L. Bylayers as Phase Transfer Agents for Nanocrystals Prepared in Nonpolar Solvents. ACS Nano 2009, 3, 2139–2146. [Google Scholar] [CrossRef] [PubMed]
- Rostami, A.; Atashkar, B.; Moradi, D. Synthesis, characterization and catalytic properties of magnetic nanoparticle supported guanidine in base catalyzed synthesis of α-hydroxyphosphonates and α-acetoxyphosphonates. Appl. Catal. A Gen. 2013, 467, 7–16. [Google Scholar] [CrossRef]
- Liu, B.; De Folter, J.W.J.; Möhwald, H. Magnetic nanoparticles-induced anisotropic shrinkage of polymer emulsion droplets. Soft Matter 2011, 7, 3744–3749. [Google Scholar] [CrossRef]
- Wang, M.; Peng, M.-L.; Cheng, W.; Cui, Y.-L.; Chen, C. A novel approach for transfering oleic acid capped Iron Oxide Nanoparticles to water phase. J. Nanosci. Nanotechnol. 2011, 11, 3688–3691. [Google Scholar] [CrossRef] [PubMed]
- Macha, S.F.; Limbach, P.A. Matrix-assisted laser desorption/ionization (MALDI) mass spectrometry of polymers. Curr. Opin. Solid State Mater. Sci. 2002, 6, 213–220. [Google Scholar] [CrossRef]
- Wu, K.J.; Odom, R.W. Characterizing synthetic polymers by MALDI MS. Anal. chem. 1998, 70, 456A–461A. [Google Scholar] [CrossRef] [PubMed]
- Davis, K.; Qi, B.; Witmer, M.; Kitchens, C.L.; Powell, B.A.; Mefford, O.T. Quantitative measurement of ligand exchange on iron oxides via radiolabeled oleic acid. Langmuir 2014, 30, 10918–10925. [Google Scholar] [CrossRef] [PubMed]
- Davis, K.; Cole, B.; Ghelardini, M.; Powell, B.A.; Mefford, O.T. Quantitative Measurement of Ligand Exchange with Small-Molecule Ligands on Iron Oxide Nanoparticles via Radioanalytical Techniques. Langmuir 2016, 32, 13716–13727. [Google Scholar] [CrossRef] [PubMed]











© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tan, L.; Liu, B.; Siemensmeyer, K.; Glebe, U.; Böker, A. Synthesis of Polystyrene-Coated Superparamagnetic and Ferromagnetic Cobalt Nanoparticles. Polymers 2018, 10, 1053. https://doi.org/10.3390/polym10101053
Tan L, Liu B, Siemensmeyer K, Glebe U, Böker A. Synthesis of Polystyrene-Coated Superparamagnetic and Ferromagnetic Cobalt Nanoparticles. Polymers. 2018; 10(10):1053. https://doi.org/10.3390/polym10101053
Chicago/Turabian StyleTan, Li, Bing Liu, Konrad Siemensmeyer, Ulrich Glebe, and Alexander Böker. 2018. "Synthesis of Polystyrene-Coated Superparamagnetic and Ferromagnetic Cobalt Nanoparticles" Polymers 10, no. 10: 1053. https://doi.org/10.3390/polym10101053