Structural, Mechanical, and Dynamical Properties of Amorphous Li2CO3 from Molecular Dynamics Simulations


Abstract
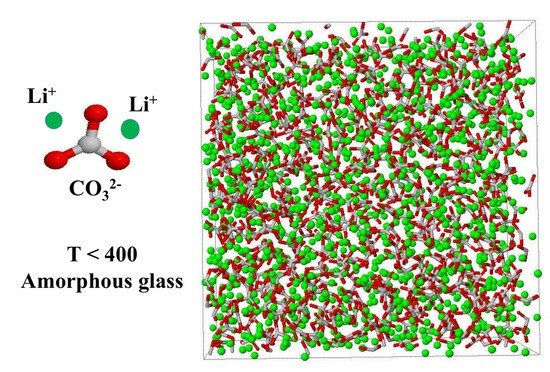
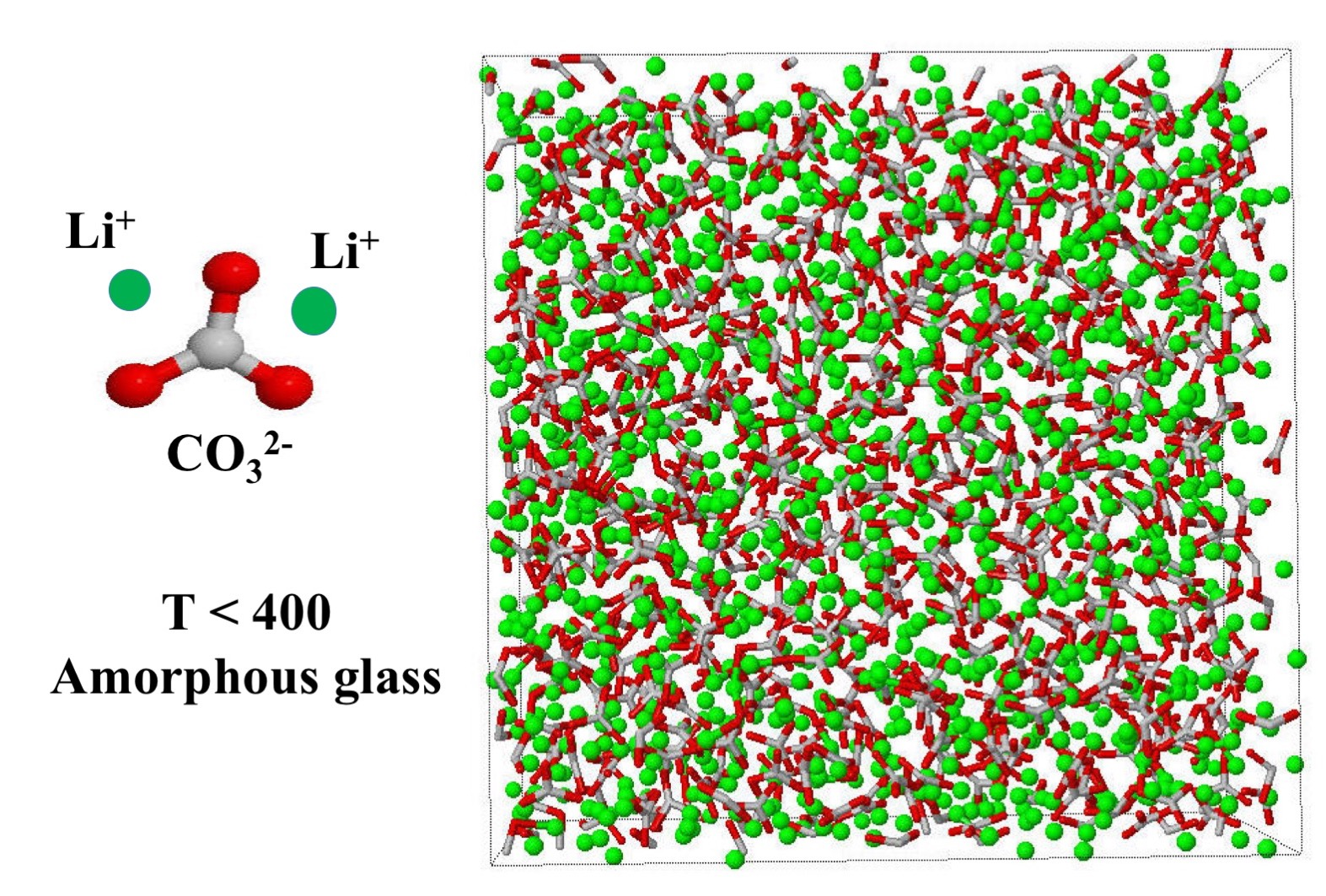
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
3. Results and Discussion
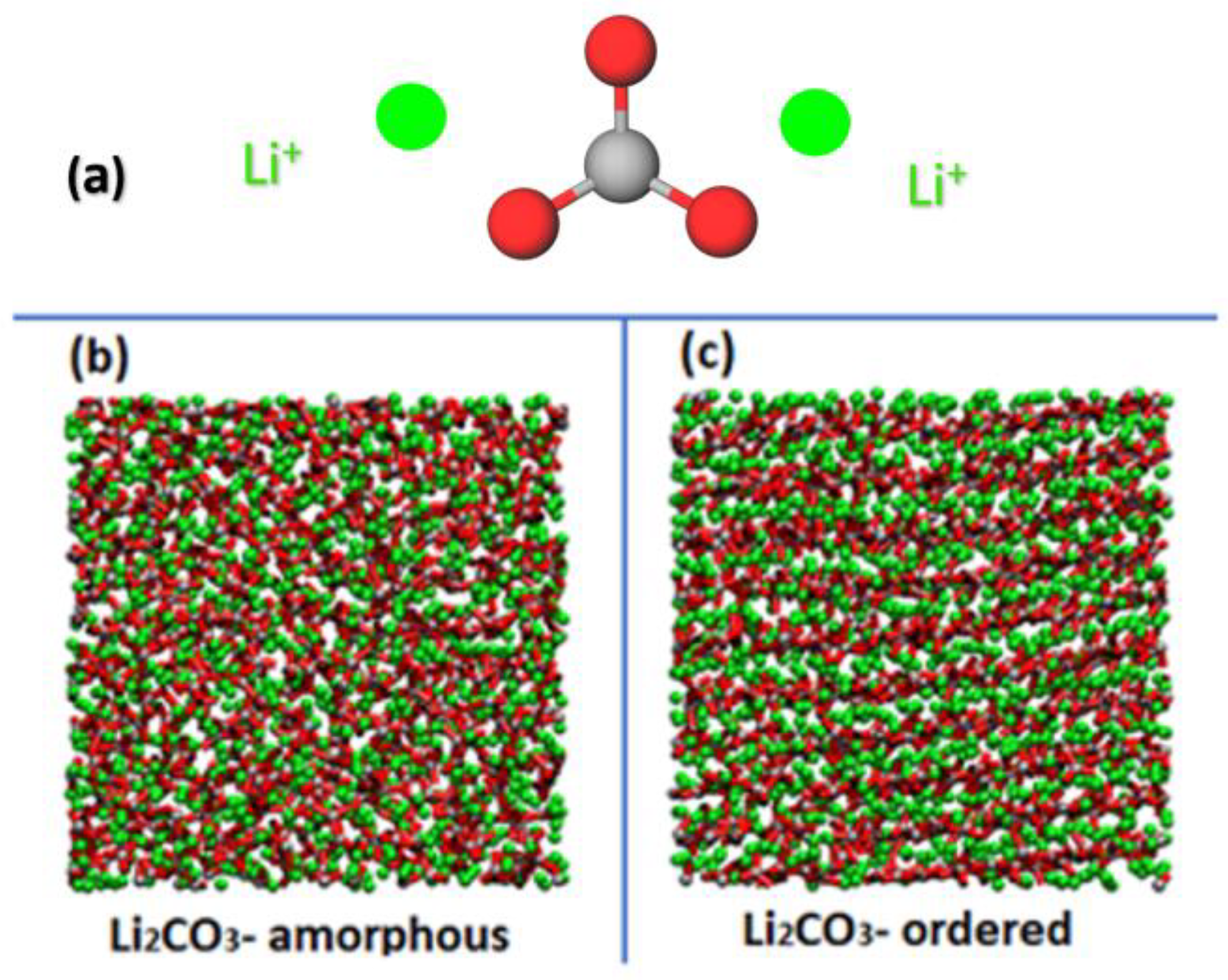
3.1. Structural Properties
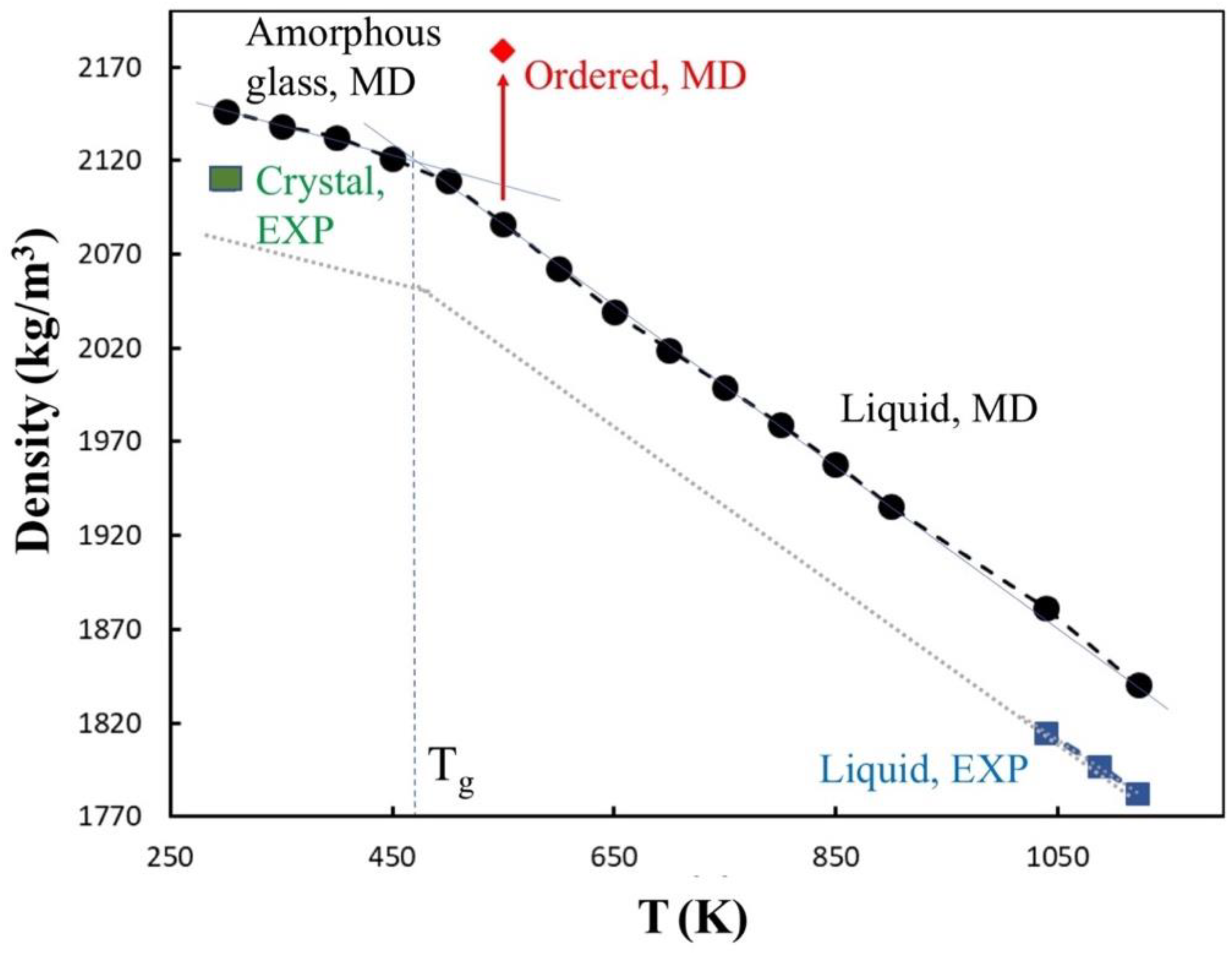
3.1.1. Density
3.1.2. Radial Distribution Functions
3.2. Dynamical Properties
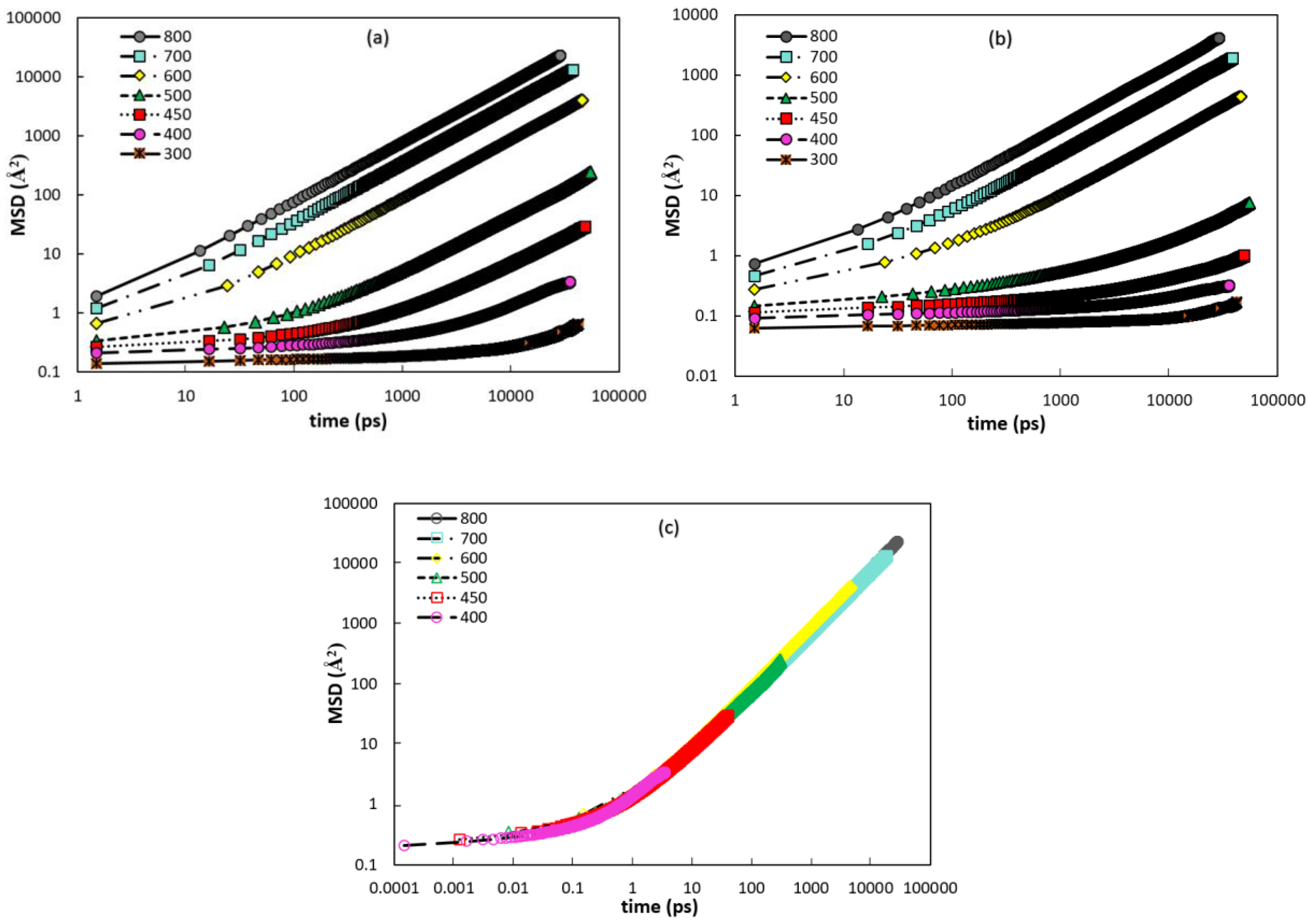
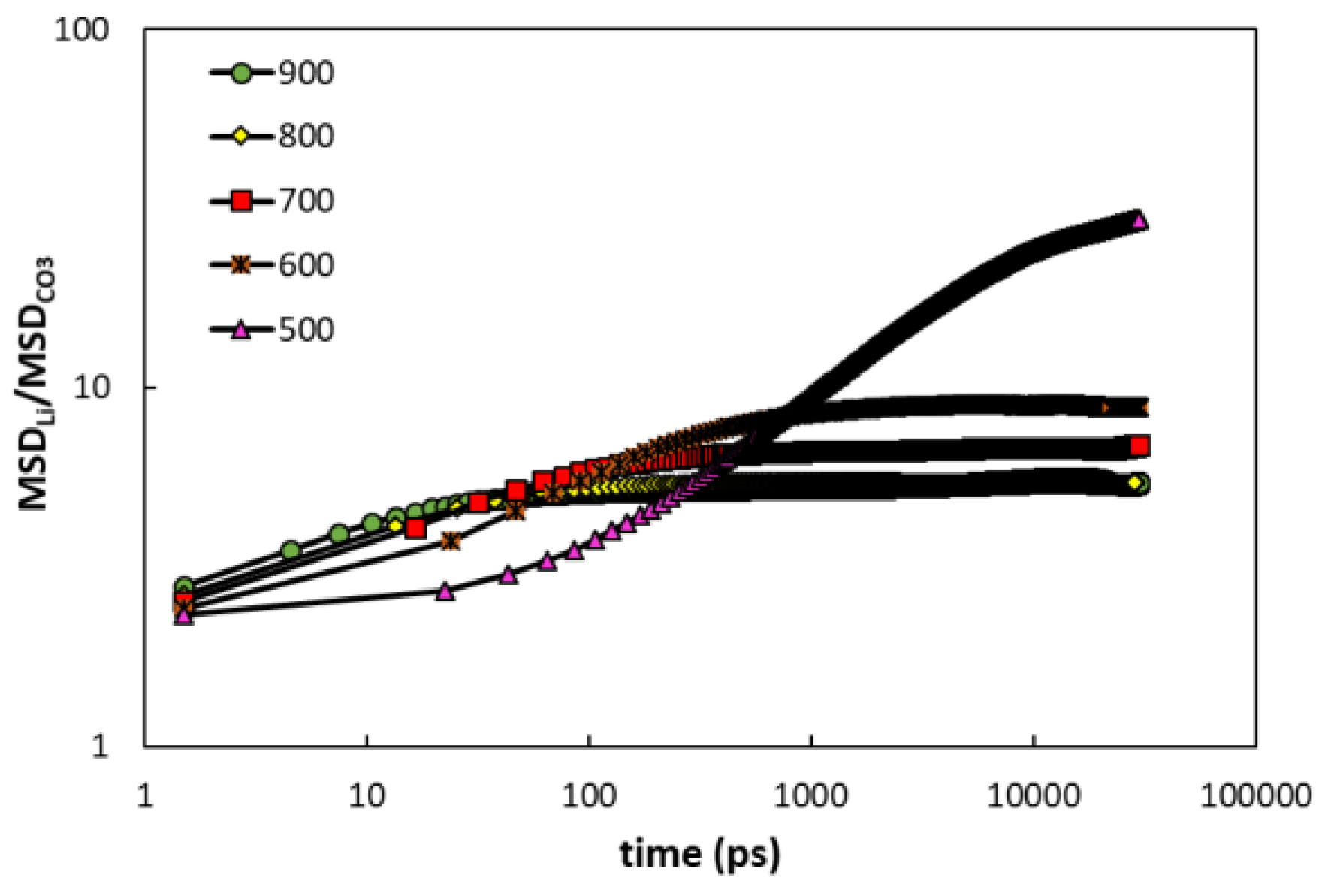
3.2.1. Mean Squared Displacements
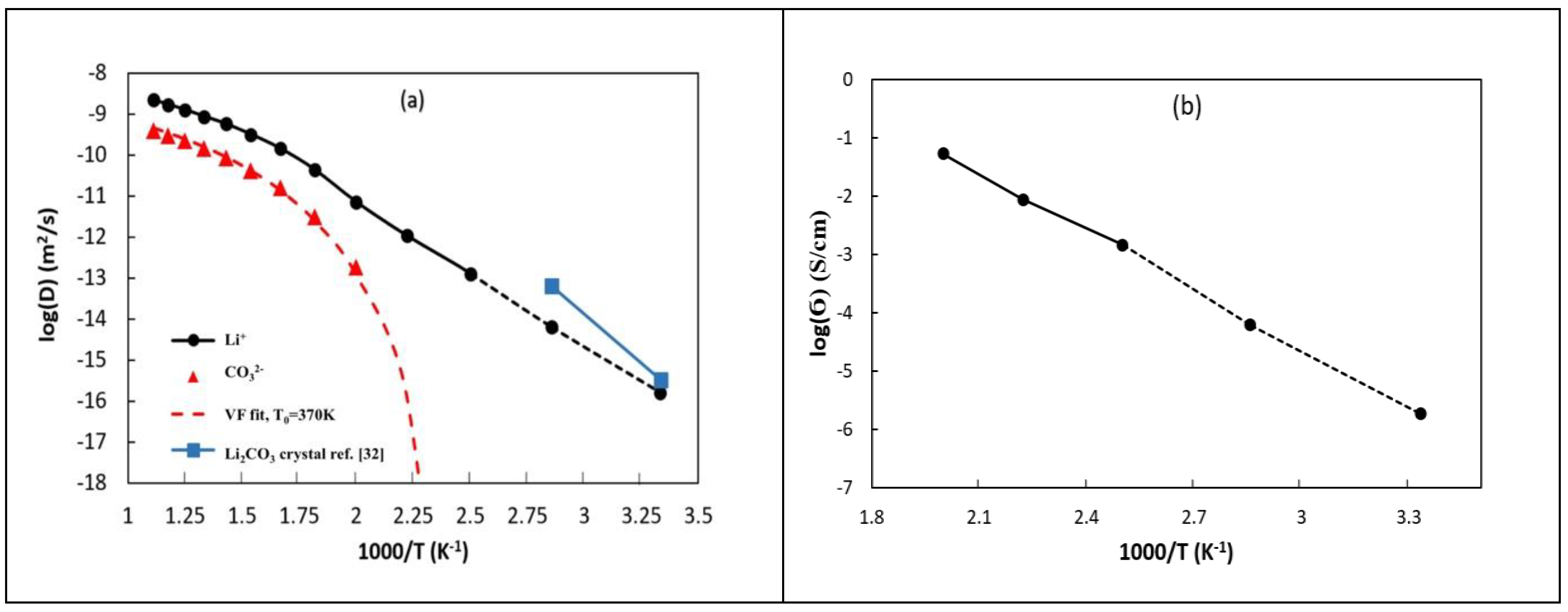
3.2.2. Self-Diffusion Coefficients
3.2.3. Ionic Conductivities
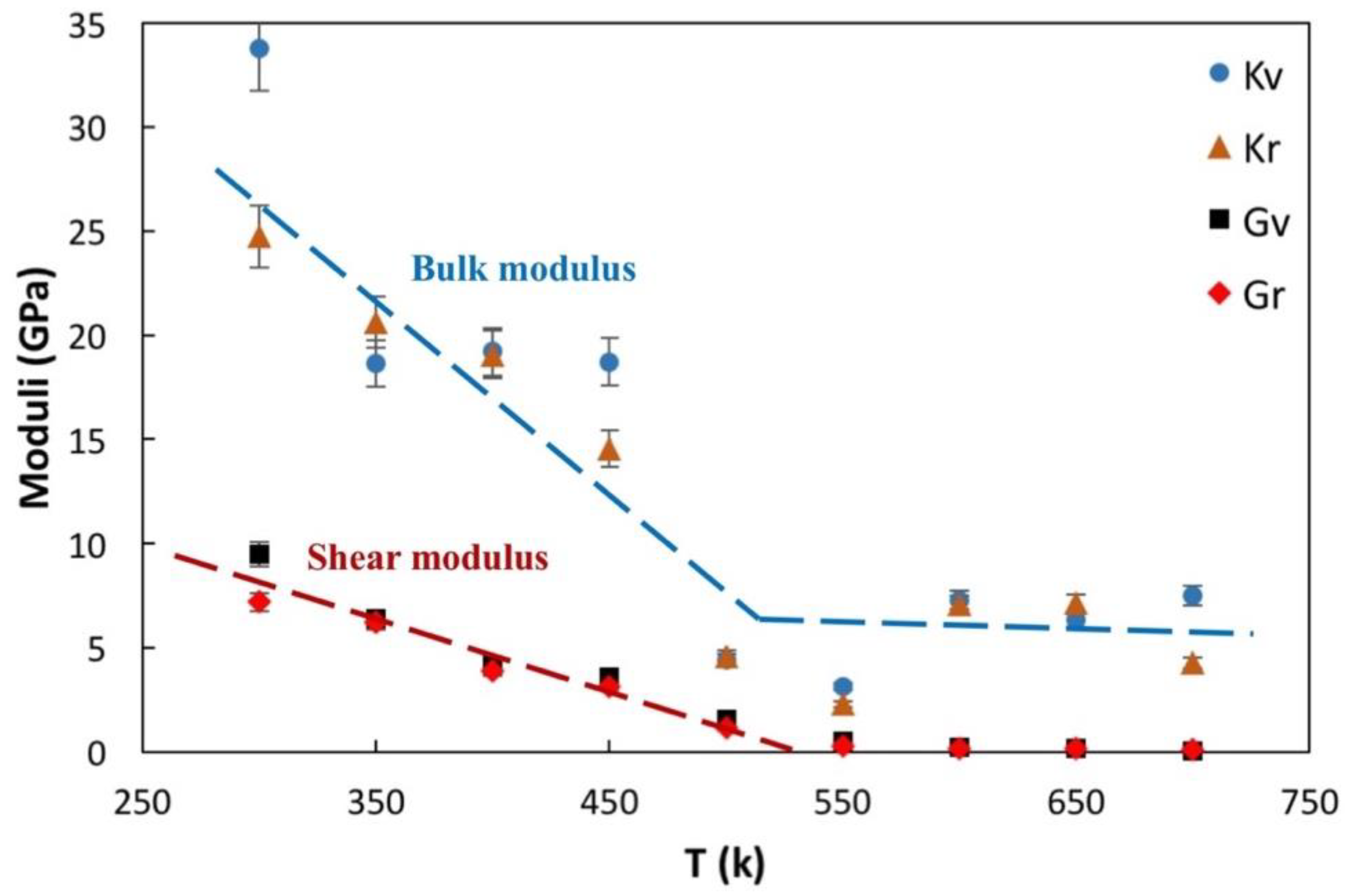
3.3. Mechanical Properties
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Shi, S.; Qi, Y.; Li, H.; Hector, L.G., Jr. Defect thermodynamics and diffusion mechanisms in Li2CO3 and implications for the solid electrolyte interphase in Li-ion batteries. J. Phys. Chem. C 2013, 117, 8579–8593. [Google Scholar] [CrossRef]
- Iddir, H.; Curtiss, L.A. Li ion diffusion mechanisms in bulk monoclinic Li2CO3 crystals from density functional studies. J. Phys. Chem. C 2010, 114, 20903–20906. [Google Scholar] [CrossRef]
- Suo, L.; Oh, D.; Lin, Y.; Zhuo, Z.; Borodin, O.; Gao, T.; Wang, F.; Kushima, A.; Wang, Z.; Kim, H.-C.; et al. How solid-electrolyte interphase forms in aqueous electrolytes. J. Am. Chem. Soc. 2017, 139, 18670–18680. [Google Scholar] [CrossRef] [PubMed]
- Bedrov, D.; Borodin, O.; Hooper, J.B. Li+ transport and mechanical properties of model solid electrolyte interphases (SEI): Insight from atomistic molecular dynamics simulations. J. Phys. Chem. C 2017, 121, 16098–16109. [Google Scholar] [CrossRef]
- Verma, P.; Maire, P.; Novák, P. A review of the features and analyses of the solid electrolyte interphase in Li-ion batteries. Electrochim. Acta 2010, 55, 6332–6341. [Google Scholar] [CrossRef]
- Wang, A.; Kadam, S.; Li, H.; Shi, S.; Qi, Y. Review on modeling of the anode solid electrolyte interphase (SEI) for lithium-ion batteries. npj Comput. Mater. 2018, 4, 15. [Google Scholar] [CrossRef]
- Aurbach, D.; Ein-Eli, Y.; Markovsky, B.; Zaban, A.; Luski, S.; Carmeli, Y.; Yamin, H. The study of electrolyte solutions based on ethylene and diethyl carbonates for rechargeable Li batteries II. Graphite electrodes. J. Electrochem. Soc. 1995, 142, 2882–2890. [Google Scholar] [CrossRef]
- Zorba, V.; Syzdek, J.; Mao, X.; Russo, R.E.; Kostecki, R. Ultrafast laser induced breakdown spectroscopy of electrode/electrolyte interfaces. Appl. Phys. Lett. 2012, 100, 234101. [Google Scholar] [CrossRef]
- Li, T.; Balbuena, P.B. Theoretical studies of the reduction of ethylene carbonate. Chem. Phys. Lett. 2000, 317, 421–429. [Google Scholar] [CrossRef]
- Peled, E.; Golodnitsky, D.; Ardel, G. Advanced model for solid electrolyte interphase electrodes in liquid and polymer electrolytes. J. Electrochem. Soc. 1997, 144, L208–L210. [Google Scholar] [CrossRef]
- Vollmer, J.M.; Curtiss, L.A.; Vissers, D.R.; Amine, K. Reduction Mechanisms of Ethylene, Propylene, and Vinylethylene Carbonates A Quantum Chemical Study. J. Electrochem. Soc. 2004, 151, A178–A183. [Google Scholar] [CrossRef]
- Leung, K.; Soto, F.; Hankins, K.; Balbuena, P.B.; Harrison, K.L. Stability of Solid Electrolyte Interphase Components on Li Metal and Reactive Anode Material Surface. J. Phys. Chem. C 2016, 120, 6302–6313. [Google Scholar] [CrossRef]
- Choi, Y.-K.; Chung, K.-I.; Kim, W.-S.; Sung, Y.-E.; Park, S.-M. Suppressive effect of Li2CO3 on initial irreversibility at carbon anode in Li-ion batteries. J. Power. Sources 2002, 104, 132–139. [Google Scholar] [CrossRef]
- Shin, J.-S.; Han, C.-H.; Jung, U.-H.; Lee, S.-I.; Kim, H.-J.; Kim, K. Effect of Li2CO3 additive on gas generation in lithium-ion batteries. J. Power Sources 2002, 109, 47–52. [Google Scholar] [CrossRef]
- Zhang, S.; Xu, K.; Jow, T. Effect of Li2CO3-coating on the performance of natural graphite in Li-ion battery. Electrochem. Commun. 2003, 5, 979–982. [Google Scholar] [CrossRef]
- Kazyak, E.; Chen, K.H.; Davis, A.L.; Yu, S.; Sanchez, A.J.; Lasso, J.; Bielinski, A.R.; Thompson, T.; Sakamoto, J.; Siegel, D.J.; et al. Atomic layer deposition and first principles modeling of glassy Li3BO3–Li2CO3 electrolytes for solid-state Li metal batteries. J. Mater. Chem. A 2018, 6, 19425–19437. [Google Scholar] [CrossRef]
- Sulaiman, M.; Rahman, A.A.; Mohamed, N.S. Sol–gel synthesis and characterization of Li2CO3–Al2O3 composite solid electrolytes. Ionics 2016, 22, 327–332. [Google Scholar] [CrossRef]
- Shang, S.-L.; Hector, L.G., Jr.; Shi, S.; Qi, Y.; Wang, Y.; Liu, Z.-K. Lattice dynamics, thermodynamics and elastic properties of monoclinic Li2CO3 from density functional theory. Acta Mater. 2012, 60, 5204–5216. [Google Scholar] [CrossRef]
- Kim, J.W.; Lee, H.G. Thermal and carbothermic decomposition of Na2CO3 and Li2CO3. Metall. Mater. Trans. B 2001, 32, 17–24. [Google Scholar] [CrossRef]
- Xu, K. Manipulating interphases in batteries. Natl. Sci. Rev. 2017, 4, 19–20. [Google Scholar] [CrossRef]
- Borodin, O.; Zhuang, G.V.; Ross, P.N.; Xu, K. Molecular dynamics simulations and experimental study of lithium ion transport in dilithium ethylene dicarbonate. J. Phys. Chem. C 2013, 117, 7433–7444. [Google Scholar] [CrossRef]
- Oldiges, K.; Diddens, D.; Ebrahiminia, M.; Hooper, J.B.; Cekic-Laskovic, I.; Heuer, A.; Bedrov, D.; Winter, M.; Brunklaus, G. Understanding transport mechanisms in ionic liquid/carbonate solvent electrolyte blends. Phys. Chem. Chem. Phys. 2018, 20, 16579–16591. [Google Scholar] [CrossRef] [PubMed]
- Martyna, G.J.; Tuckerman, M.E.; Tobias, D.J.; Klein, M.L. Explicit reversible integrators for extended systems dynamics. Mol. Phys. 1996, 87, 1117–1157. [Google Scholar] [CrossRef]
- Bedrov, D.; Borodin, O.; Smith, G.D.; Sewell, T.D.; Dattelbaum, D.M.; Stevens, L.L. A molecular dynamics simulation study of crystalline 1, 3, 5-triamino-2, 4, 6-trinitrobenzene as a function of pressure and temperature. J. Chem. Phys. 2009, 131, 224703. [Google Scholar] [CrossRef] [PubMed]
- Hooper, J.B.; Borodin, O.; Schneider, S. Insight into hydrazinium nitrates, azides, dicyanamide, and 5-azidotetrazolate ionic materials from simulations and experiments. J. Phys. Chem. B 2011, 115, 13578–13592. [Google Scholar] [CrossRef] [PubMed]
- Sewell, T.D.; Menikoff, R.; Bedrov, D.; Smith, G.D. A molecular dynamics simulation study of elastic properties of HMX. J. Chem. Phys. 2003, 119, 7417–7426. [Google Scholar] [CrossRef]
- Liu, Q.; Lange, R.A. New density measurements on carbonate liquids and the partial molar volume of the CaCO3 component. Mineral. Petrol. 2003, 146, 370–381. [Google Scholar] [CrossRef]
- Kojima, T.; Miyazaki, Y.; Nomura, K.; Tanimoto, K. Density, Molar Volume, and Surface Tension of Molten Li2CO3 Na2CO3 and Li2CO3 K2CO3 Containing Alkaline Earth (Ca, Sr, and Ba) Carbonates. J. Electrochem. Soc. 2003, 150, E535–E542. [Google Scholar] [CrossRef]
- Lide, D.R. Physical Constant of Inorganic Compound. In Handbook of Chemistry and Physics; CRC Press: Boca Raton, FL, USA, 2005. [Google Scholar]
- Islam, M.M.; Ostadhossein, A.; Borodin, O.; Yeates, A.T.; Tipton, W.W.; Hennig, R.G.; Kumar, N.; van Duin, A.C.T. ReaxFF molecular dynamics simulations on lithiated sulfur cathode materials. Phys. Chem. Chem. Phys. 2015, 17, 3383–3393. [Google Scholar] [CrossRef] [PubMed]
- Kohara, S.; Badyal, Y.S.; Koura, N.; Idemoto, Y.; Takahashi, S.; Curtiss, L.A.; Saboungi, M.L. The structure of molten alkali carbonates studied by neutron diffraction and ab initio calculations. J. Phys. Condens. Matter. 1998, 20, 3301. [Google Scholar] [CrossRef]
- Benitez, L.; Seminario, J.M. Ion diffusivity through the solid electrolyte interphase in lithium-ion batteries. J. Electrochem. Soc. 2017, 164, E3159–E3170. [Google Scholar] [CrossRef]
- Parrinello, M.; Rahman, A. Strain fluctuations and elastic constants. J. Chem. Phys. 1982, 76, 2662–2666. [Google Scholar] [CrossRef]
- Tsai, S.W. AFML Techncal Report No. AFML-TR-66-149; Air Force Materials Laboratory, Research and Technology Division: USA, 1966. [Google Scholar]
- Voigt, W. Theoretical studies of the elastic behaviour of crystals. Presented at the session of the Royal Society of Science. Available online: http://www.neo-classical-physics.info/uploads/3/4/3/6/34363841/voigt_-_elasticity_of_crystals_-_i.pdf (accessed on 18 December 2018).
- Reuss, A. Berechnung der fließgrenze von mischkristallen auf grund der plastizitätsbedingung für einkristalle. J. Appl. Math. Mech./ZAMM 1929, 9, 49–58. [Google Scholar] [CrossRef]
- Hill, R. The elastic behaviour of a crystalline aggregate. Proc. Phys. Soc. A 1952, 65, 349. [Google Scholar] [CrossRef]
- Monroe, C.; Newman, J. The impact of elastic deformation on deposition kinetics at lithium/polymer interfaces. J. Electrochem. Soc. 2005, 152, A396–A404. [Google Scholar] [CrossRef]







© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ebrahiminia, M.; Hooper, J.B.; Bedrov, D. Structural, Mechanical, and Dynamical Properties of Amorphous Li2CO3 from Molecular Dynamics Simulations. Crystals 2018, 8, 473. https://doi.org/10.3390/cryst8120473
Ebrahiminia M, Hooper JB, Bedrov D. Structural, Mechanical, and Dynamical Properties of Amorphous Li2CO3 from Molecular Dynamics Simulations. Crystals. 2018; 8(12):473. https://doi.org/10.3390/cryst8120473
Chicago/Turabian StyleEbrahiminia, Mahsa, Justin B. Hooper, and Dmitry Bedrov. 2018. "Structural, Mechanical, and Dynamical Properties of Amorphous Li2CO3 from Molecular Dynamics Simulations" Crystals 8, no. 12: 473. https://doi.org/10.3390/cryst8120473