2. Distributed Atomic and Functional-Group Polarizabilities
In order to identify the most important atoms and functional groups that determine the optical properties of a molecule or a crystalline material, we need a partitioning of the electrostatic moments and (hyper)polarizabilities into atomic contributions. In experimental electron-density determination, a formalism based on pseudo-atoms is widely adopted, which enables the refinement of electron-density models against measured structure-factor amplitudes [
5]. In principle, the refined coefficients may be used to compute atomic electrostatic moments. However, there are two main limitations: (a) the multipole expansion is a fitting procedure that therefore returns only approximated quantities; and (b) the atomic multipole parameters may strongly correlate within a refinement procedure, therefore while the total electron density is quite accurately reconstructed, the atomic partitioning may be biased. A better estimation of atomic moments derives from space-partitioning of the total electron density reconstructed from the refined coefficients of the multipole expansion. In fact, the total electron density is less affected by the correlation among parameters of the multipolar expansion. For the partition, one can use the quantum theory of atoms in molecules (QTAIM) [
6], which is more generally applicable to a theoretical electron density as well. An advantage of QTAIM is that the partitioning is
exact (meaning that the sum of atomic contributions exactly reconstruct the total property) and the atomic basins are discrete, making it easier the definition of an important quantity like the charge transfer (see
Supplementary Materials). Furthermore, QTAIM-basins are typically very well transferable among systems, thus allowing their accurate estimation in small, model molecules and exportation to larger ones. Similar schemes could be for example the Voronoi polyhedra or the Hirshfeld surface partitioning. Both are exclusive partitioning of the space, although the latter leaves some small volumes unassigned [
7]. An alternative approach is the Hirshfeld atom: here the partitioning is complete, but the atomic basins overlap [
8], which may create problems for assigning correctly the interatomic charge transfer.
Some Hilbert-space partitionings, such as Stone’s [
9], could also be used to obtain atomic or functional-group moments and (hyper)polarizabilities. However, with these methods, the partitioning suffers from being largely dependent on the atomic basis-sets and from a limited transferability. Notably, the computationally cheap Mulliken’s partition [
10] holds for atomic charges only. Although these can reconstruct a molecular dipole, the lack of terms describing the atomic intrinsic polarization (see
Supplementary Materials) does not enable computing a fundamental part of the atomic polarizability.
Some partitioning schemes have been applied to estimate linear optical properties of atoms and functional groups in molecules or crystals. Several approaches have been proposed for the calculation of
distributed atomic polarizabilities, i.e., the atomic polarizability tensors within a molecule or molecular aggregate (for a recent account, the reader is referred to [
4] and references therein). Among those, Bader and coworkers developed QTAIM-partitioned polarizabilities and used them to evaluate intermolecular interaction energies and transferability of electric properties [
11]. Keith’s generalization of Bader’s method removed the origin dependence from the QTAIM definition of atomic dipoles and polarizabilities, thus making them perfectly transferable to other systems [
12]. Within this approach, the component
of the atomic first-order polarizability tensor can be simply evaluated through the numerical derivative of the corresponding atomic dipole moments, with respect to an externally applied electric field:
where
is the dipole moment component of the atomic basin
along the
j direction computed with an electric field applied along the direction
i. Keith’s approach has been recently modified and implemented in a program called
PolaBer [
13], used to calculate the distributed polarizability tensors reported in this paper (for details of the implementation, as well as for the methodological procedures used in this work, see
Supporting Information). The total, molecular or crystal polarizability can be straightforwardly obtained through summation of the atomic counterparts, as typical for any molecular property estimated using QTAIM. Because the polarizabilities have dimensions of volume, atomic and molecular tensors can be visualized as ellipsoids, see, for example,
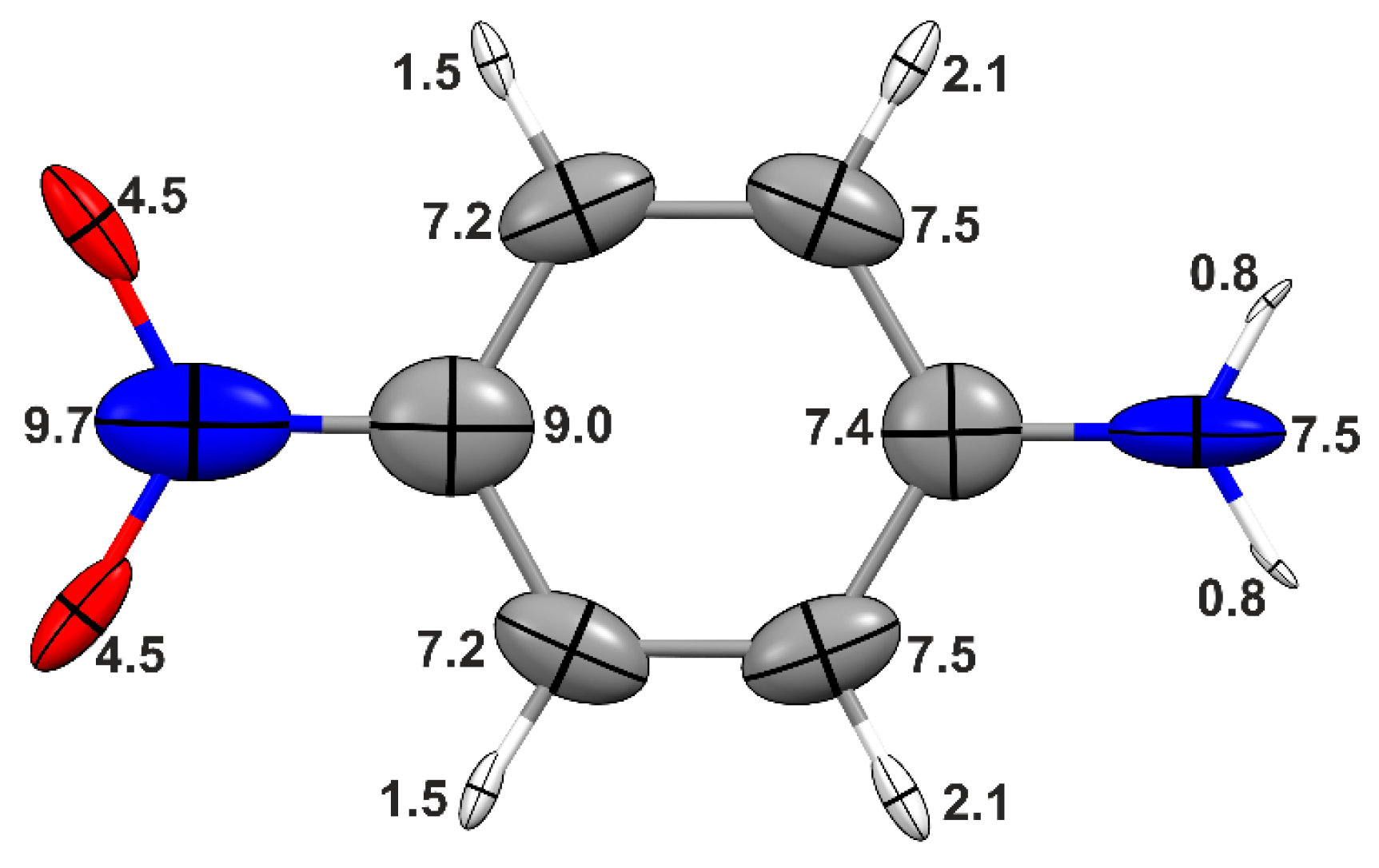
Figure 1, which shows
p-nitroaniline, calculated at the CAM-B3LYP/cc-pVDZ level of theory. Most of the calculations in this work were performed using the modest-size cc-pVDZ basis-set. Even though augmentation with diffuse functions is highly desirable to accurately estimate electric moments and polarizabilities of molecules in an infinitely diluted gas, these functions are less relevant for estimating properties in aggregates or crystals, due to the existence of a “basis-set superposition” effect that takes place among basis-sets of vicinal groups or molecules in aggregation, see for example [
14].
Otero
et al. [
15] criticized the use of Equation (1) because the ground-state molecular electron density
and the corresponding field-perturbed densities
are “independently” partitioned into their atomic contributions, without guaranteeing that the corresponding atomic basins are kept constant in the process. They developed, instead, a methodology to partition the polarizability distribution
using Hirshfeld scheme.
enables visualizing regions of negative polarizability in a molecule,
i.e., regions that would respond counterintuitively to an applied field (with negative charges moving in the direction of the negative field).
The criticism of Otero
et al. [
15] is, however, not fully correct. First of all, the numerical approach in Equation (1), in the limit of an infinitesimally small field, gives the exact definition of the dipolar electron density derivative that coincides with the analytical definition. Therefore, there is no bias in applying Equation (1) and the numerical solution is used for the sake of simplicity in the complex step of the atomic basin integration and the successive derivation with the field. If the values obtained through the numerical approach do not sum up perfectly to the total molecular polarizability (typically in the range of 0.1%), this can be easily overcome by using more precise integration inside the atomic basins and smaller electric fields. The integration is anyway the most crucial step, because of the difficult definition of QTAIM atomic basins. The fact that the atomic tensors are not symmetrical, instead, depends primarily on the fact that the atomic basins are themselves not symmetrical, which does not enable Equation (1) to be invariant with respect to exchange of
i and
j. Noteworthy, this is true with any discrete partitioning (e.g., Voronoi or Hirshfeld surface), unless the atom lies on a symmetric site. It holds also for the Hirshfeld atomic partitioning, because, although the volume in which atoms are integrated is infinite, the weight function that defines each atom is not symmetric (again unless the atom sits on a symmetric position). Therefore, the tensor symmetrization is a necessary step, used for visualizing the atomic polarizability ellipsoid. The non-symmetrized tensors would be the exact QTAIM-derived atomic tensors, but would also be more difficult to use. The tensor symmetrization has two important properties: (a) the isotropic atomic polarizability is invariant (because only the out of diagonal components are touched); and (b) the sum of the atomic tensors gives anyway the total (symmetric and exact) molecular polarizability, because the anti-symmetric part of the atomic tensors mutually cancel each other. Otero
et al. [
15] also claimed that in the calculation of integrated atomic polarizabilities, the regions of negative
are “explicitly omitted”. On the contrary, the integration inside an atomic basin implies that regions of negative polarizability be overwhelmed (not omitted) by those of positive polarizability, producing overall the expected result that on average the electron density responds intuitively to the applied field. Interestingly, however, the remark of Otero
et al. [
15] raises the question whether some atoms in a molecule may in principle have negative atomic tensors. Albeit possible (and anyway partitioning dependent), this is unlikely to occur for all atoms with a core, which necessarily implies a sufficiently large and almost symmetric basin. Therefore, the only atoms that may display a negative polarizability are H atoms, especially if involved in strong HB (where they exhibit small and irregular atomic basins).
Despite the above discussed criticism, it has been demonstrated that QTAIM distributed polarizabilities obtained using
PolaBer are remarkably exportable among some series of molecules, including amino acids and organic optical materials [
16]. Databanks of atomic and functional-group polarizabilities have been created and used to estimate the refractivity of a few materials [
4]. Furthermore, this approach has been successfully applied to understand the origin of the refractive indices of molecular crystals in terms of their most fundamental building blocks,
i.e., atoms and functional groups [
17], as well as to quantify the effect of hydrogen bonding on their optical behavior (see next section).
3. From Functional Groups and Molecules to Crystals
High-resolution X-ray diffraction is an established technique for determination of molecular electrostatic moments of atoms and molecules in crystals, with the advantage of providing not only magnitudes, but also all their tensor components. Some time ago, Fkyerat
et al. [
18,
19] and Hamzaoui
et al. [
20,
21] proposed a correlation between the molecular first-order polarizability
α and first hyperpolarizability
β with, respectively, the quadrupole and octupole electrostatic moments, derivable from the X-ray experiments. However, application of this method to organic non-linear optical materials revealed that many components of
α and
β tensors, calculated from X-ray diffraction, differ by more than an order of magnitude from theoretical results. Indeed, it was later shown that the one-electron density
, obtained from the usual multipolar pseudo-atom formalisms, does not yield accurate response properties because electronic correlation is included only partially [
22]. Instead, Jayatilaka and Cole [
23] have pursued an X-ray constrained wavefunction approach to derive much more accurate
distributions for a few optical materials, including a metal-organic non-linear optical compound. The “experimental” wavefunctions yielded remarkably accurate electric properties, indicating the possibility to use X-ray constrained molecular orbitals for engineering this kind of materials. In the case a scheme is further assumed to localize the constrained canonical orbitals in particular atoms or functional groups [
24], they can be exported even to much larger systems, thus allowing an inexpensive, though accurate, prediction of their electric properties as well, which is currently challenging for both theory and experiment.
The estimation of electrical properties from X-ray diffraction or fully periodic quantum-mechanical calculations [
25] is indeed highly desirable, as these techniques inherently account for the entire crystal field. However, the need of high-quality single-crystals and the high computational costs, respectively, often hamper the application of these methods for large systems. Therefore, calculations on isolated molecules have been extensively applied to estimate electrostatic moments and (hyper)polarizabilities [
3]. This requires including an empirical perturbation of the molecular electron density mimicking the crystal electric field. In fact, if the molecular electrostatic moments are calculated in an infinitely diluted gas, the agreement with experimental results in crystals (or with periodic calculations) are obviously poor.
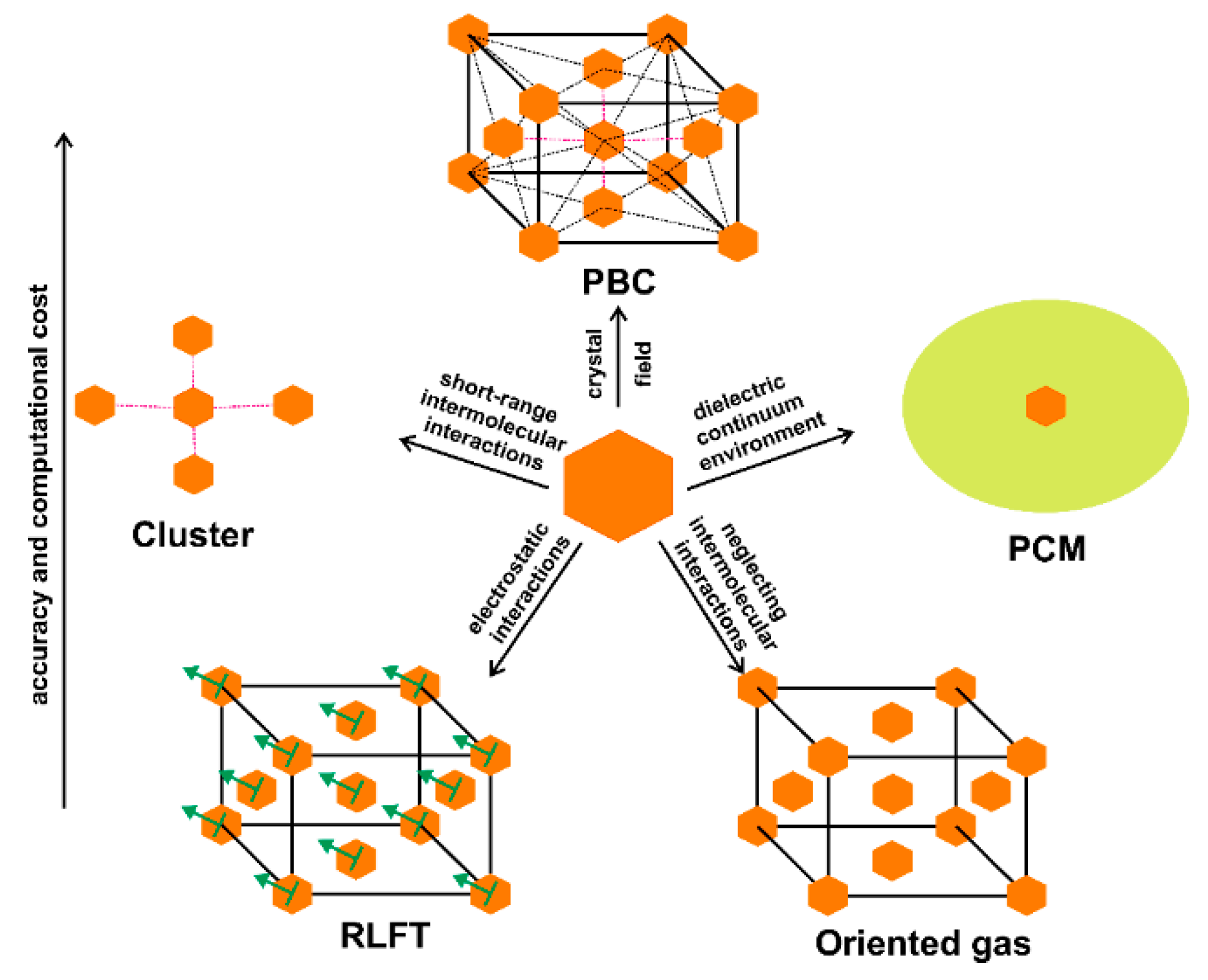
The big challenge in computational chemistry is how to simulate the crystal electric field at the lowest computational costs. We should first of all consider that molecular electric moments are typically affected by short- and long-range effects [
4]. Short-range effects are, for example, hydrogen bonds or other medium-strength intermolecular interactions. They may cause changes of the electron distribution and the molecular geometry as well, but they are possible only within the first coordination sphere around a molecule, therefore they are few. On the other hand, long-range effects, due mainly to electrostatic forces produced by interaction between electron-density clouds, are weaker (as their magnitude decrease as a power function of the intermolecular distance), but because they are potentially infinite in number their contribution may be of significance.
Hydrogen bonds tend to line up the molecules in order to minimize the energy [
2,
17]. This often, though not always, enhances their dipole moments due to induced polarization of the electron-density distributions [
26]. This
dipole moment enhancement observed when going from a single-molecule to an aggregated environment has been investigated in quite some detail for 2-methyl-4-nitroaniline, one of the molecular prototypes for linear and non-linear optical materials. A careful diffraction study by Spackman [
27] reported a significant enhancement of 30%–40%, nevertheless smaller than that reported earlier by Howard
et al. [
28]. Spackman [
26] has investigated more systematically a large number of crystals, calculating dipole enhancements up to
ca. 30% for hydrogen-bonded species, whereas many diffraction experiments result in enhancements greater than 100%. However, it is recognized that molecular moments are highly dependent on the multipole model fitted against the X-ray diffracted intensities, and several studies reporting incredibly large enhancements were performed treating the thermal motion of hydrogen atoms as isotropic and without incorporating neutron diffraction estimates of X–H distances. Molecular dipole moments may be very difficult to estimate from experimental multipolar expansion, because they strongly depend on the contraction/expansion of atomic shells, often sensitive to even minor systematic effects in the datasets. Moreover, in non-centrosymmetric crystals, special attention is necessary to avoid model ambiguities, which often affect the estimation of molecular dipole moment. Last but not least, it should also be considered that some supramolecular packings may produce dipole depletion instead of enhancement.
The redistribution of electronic charge that molecules undergo upon aggregation has also important consequences for the estimation of optical properties. Studies on organic non-linear optical materials revealed that the hyperpolarizability of a molecule typically grows by a factor of three when a hydrogen-bonded cluster is considered in its surrounding [
29]. Instead, the first-order polarizability
α is much less susceptible, thus being determined to a great extent by the intramolecular connectivity (the chemical bonds) rather than the intermolecular forces. As an example, we consider the atomic polarizabilities corresponding to a head-to-tail aggregation of two
p-nitroaniline molecules (see
Figure 2), and compare these values with those of
Figure 1. The moderate-to-weak hydrogen bond characterizing this system increases the polarizability of the donor (–NH
2) and acceptor (–NO
2) functional groups by 7% and 16%, respectively, whereas the polarizability of the phenyl ring enhances only slightly (approximately 1%). As shown in
Table 1, the total molecular polarizability does not increase dramatically because of this interaction. In a real crystal, however, many more hydrogen bonds occur coupled with long-range interactions (electrostatics only). All these smaller contributions eventually sum up leading to a significant increase. Indeed, the molecular polarizability calculated under periodic boundary conditions (PBC) is much larger than that for the molecules composing the dimer (or for any other finite aggregate). As also shown in
Table 1, the hydrogen bond has a significant effect on the polarizability anisotropy
, a quantity directly related to the birefringence of a material.
As anticipated, calculations under periodic boundary conditions are in principle the correct approaches to model the crystalline effects, and therefore, to accurately estimate electrostatic and response properties of molecules in crystals because they might be dramatically influenced by intermolecular interactions. However, some problems affect the crystal-orbital or plane-wave-based methods: (a) the amount of electronic correlation that one can introduce is limited; (b) for crystal orbitals, convergence often fails when Bloch-type functions use diffuse atomic basis functions; and (c) plane-wave calculations, instead, exclude localized core-orbital functions and therefore their contribution to the properties, which can be quite substantial for hyperpolarizabilities, for example. In order to overcome these drawbacks, the so-called
supermolecule or
cluster method was proposed [
3]. Within this approach, the properties of interest of several interacting molecules are evaluated as a whole, just like in standard molecular calculations. The goal is then extracting the properties featured by the “central” molecule around which the cluster is constructed. In fact, by comparing the electric properties of an isolated molecule and that of the molecular cluster, one gains insight into the role of short and medium-range intermolecular interactions, crucial for the design of optical materials [
4]. However, this approach includes only partially the crystal field effects, because long-range interactions are not accounted. Furthermore, as the choice of the molecular aggregate is usually not unique, a computationally costly pre-screening of at least a few clusters is typically necessary [
30].
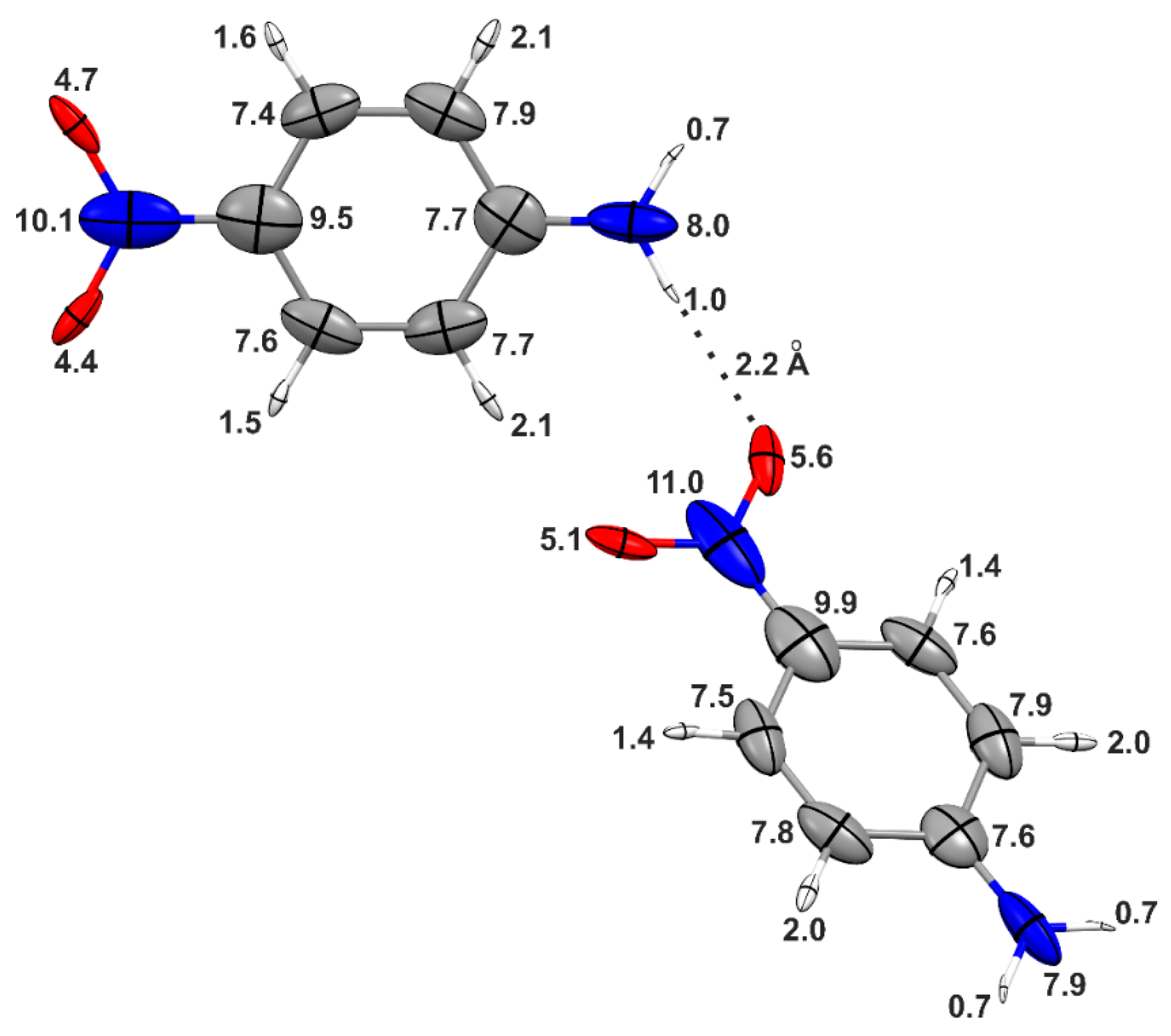
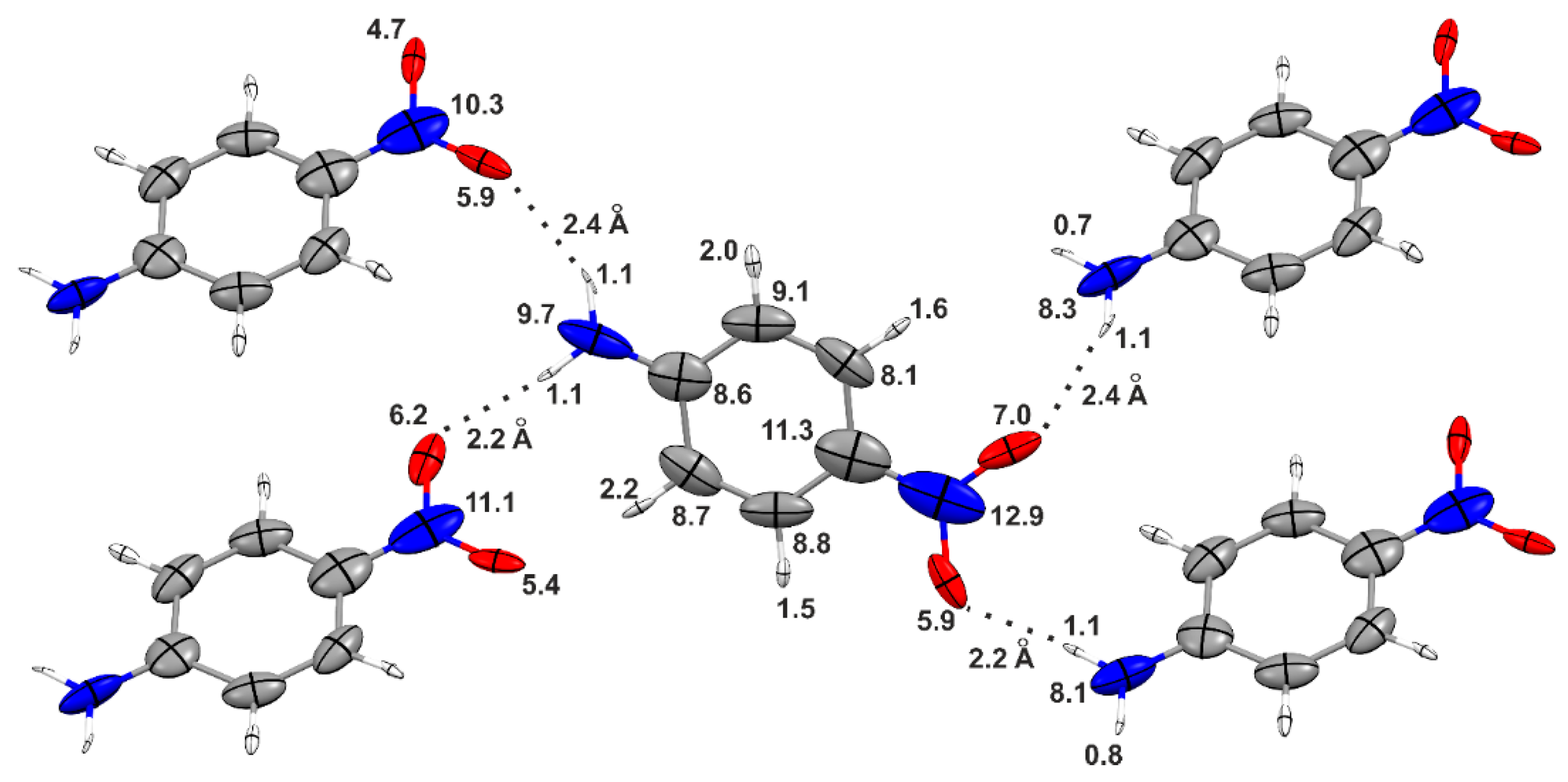
Table 1 summarizes the polarizability of
p-nitroaniline, as obtained from different aggregates constructed in order to progressively saturate the hydrogen-bond sites of the “central” molecule (see
Figure 3 and
Figure 4). When going from the monomer to the trimer, the molecular isotropic polarizability increases by 12%, and a further enhancement of 9% is observed for the central molecule in the pentamer. Although the latter corresponds to a “hydrogen-bond saturation”, this by no means indicates convergence of the polarizability because, as previously mentioned, long-range electrostatic interactions may still be of significance. Noteworthy, the polarizabilities of the hydrogen-bonded donor and acceptor groups in the central molecule of the pentamer are up to 30% larger than in the isolated molecule, whereas the enhancements in the phenyl rings are negligible. These results show that the distortions caused by the hydrogen bonds are largely localized in the functional groups directly involved in the bonding.
The cluster approach has been previously used to investigate the effect of hydrogen-bond formation on the (hyper)polarizabilities of a few other organic materials. For example, Balakina and coworkers [
29,
31], studied theoretically a series of non-linear optical chromophores and demonstrated that the formation of weak to moderate N–H···O bonds (with donor-acceptor distances in the range 2.8–3.5 Å) results in an increment of only 10%–15% of the molecular polarizability
α, whereas the first hyperpolarizability
β may increase up to three times. It is anyway noteworthy that even small changes in
α will significantly affect the calculation of the electric susceptibility through a lattice summation.
The importance of explicitly taking into account hydrogen bonds in the estimations of optical properties becomes rather evident when the supermolecule approach is compared with calculations of single-molecules embedded in a polarizable continuum dielectric medium (PCM). This approach somewhat introduces averaged electrostatic interactions, improving calculations of isolated molecules, but it is not capable of predicting the correct polarization of the electron density caused by short-range interactions with partial covalent component, such as hydrogen bonds. Thus, the PCM approach often underestimates the (hyper)polarizabilities. Nevertheless, we are currently working [
32] on a semi-continuum method, which combines PCM with calculations on a moderately large cluster, that can be quite effective to estimate optical properties of organic and metal-organic materials.
Although many studies investigated the effect of hydrogen bonding on QTAIM topological parameters, not much work has been devoted to study such influence on the corresponding atomic and functional-group polarizabilities. We have recently investigated many aggregates of amino acids in their zwitterionic configuration with the purpose to quantify the changes of
α for the hydrogen-bond donor (–NH
3+) and acceptor (–COO
−) functional groups [
4,
17]. While the isotropic polarizabilities of the groups do not change more than 10% upon hydrogen-bond formation, the tensors are quite significantly reoriented towards the hydrogen-bond direction. This means that these interactions may be useful for controlling the anisotropy of crystalline optical properties, rather than enhancing them. In fact, the anisotropy of the functional-group tensors was found to increase significantly (20%–30%) upon the formation of hydrogen bonds.
An alternative strategy to include crystal field effects in a single-molecule calculation or long-range effects in a molecular aggregate, is firstly to construct an
oriented-gas model, one in which non-interacting identical molecules or aggregates lie side-by-side and the solid-state properties are simply appropriate combinations of the molecular ones. Afterwards, the effects of the surroundings are semi-empirically approximated using local-field factors [
33]. Since molecular crystals feature in general only non-covalent intermolecular interactions, classical electrostatic models have been successfully adopted to estimate dipole moment enhancements and, ultimately, the electric response properties of crystals starting from a simple gas-phase calculation [
30].
For the purpose of defining the local-field factor, a crystal may be represented as an array of equal dipoles distributed over a space lattice. A molecule embedded in the crystal experiences a local-electric field
that, in general, is a sum of any externally applied field and the internal field that results from the dipole moments of all other molecules in the unit cell. For relatively small values of
, the dipole moment
induced at a particular molecule
k depends only on its first-order polarizability, and can be computed as:
The dipole moment of the molecule embedded in a crystal or molecular cluster can be estimated as a simple sum of its permanent dipole moment
, calculated in gas-phase, with the induced dipole
. Therefore, this approach allows the estimation of “in-crystal” dipole moments from the knowledge of the polarizability of the constituent molecules and the symmetry operations used to construct the aggregate. This formalism is usually called
rigorous local field theory (RLFT) [
34,
35], and can be straightforwardly extended to compute atomic and functional-group induced dipole moments
in the
k molecule of the crystal, provided that a partitioning scheme is used to calculate
and
. In this case, the
molecular point-dipole realization of RLFT,
i.e., the one in which each molecule is considered as a point dipole (RLFT1), would be replaced by a
distributed atomic or functional-group point-dipole treatment, in which
n atomic dipoles are distributed over the molecule (RLFT
n). The relative accuracy of these approximations depends on the size and shape of the molecules. For small compounds, such as urea or benzene [
34], RLFT1 and RLFT
n do not differ substantially, whereas RLFT
n is typically more accurate to describe the anisotropies of larger systems, as shown for m-nitroaniline [
35].
The first-order polarizabilities of the molecule embedded in its environment can be calculated through numerical differentiation of the induced dipole moments estimated through RLFT. Thus, the components
of the polarizability calculated for the molecule in isolation are perturbed and become
in the crystal [
4]. Analogously to Equation (1),
can be computed as:
Although the local electric field of Equation (3) is just a zero-order approximation, only few works have attempted to iterate the process, using the dipole moment of the embedded molecule to compute an improved approximation to the electric field [
4,
26].
Our investigations on RLFT applied to amino acid clusters [
4] suggested that in case extreme accuracy is desired, a full
ab-initio treatment of the aggregate is necessary in order to satisfactory estimate functional-group or molecular polarizabilities because the semi-empirical approaches, although sometimes able to accurately predict dipole moment enhancements, are not capable to take into account the “re-accommodation” of the molecular electron density due to the interactions within the first coordination sphere around a molecule. The presence of the molecular vicinity manifests itself not only in terms of the polarization caused by the strongest intermolecular interactions, and of the charge-transfer along the hydrogen-bond directions, but also in terms of a volume contraction that the molecules undergo when going from isolation to the molecular aggregate. We are currently performing work in these directions, but RLFT still remains to be satisfactorily corrected for these factors.
Within RLFT, the first-order electric susceptibility tensor
of a molecular crystal can simply be calculated from the
tensors by applying well-known lattice-summation schemes [
36,
37]. The permittivity tensor is then obtained as
and the three crystalline refractive indices
can be calculated from the eigenvalues
of the permittivity,
.
Table 2 summarizes the results for
p-nitroaniline by comparing RLFT (within a molecular point-dipole implementation) against the simple oriented-gas model, a coupled-perturbed Kohn–Sham wave function using PBC, and a few estimations using intermediate levels of approximation (namely, X-ray constrained Hartree-Fock wavefunction, and an oriented-gas model using the molecule extracted from the pentamer), as well as with experimental values. Noteworthy, the experimental refractive indices are larger than all theoretically predictions because the finite frequency at which they are measured increases the refractivity with respect to the calculations performed with non-oscillating fields. An extrapolation to zero frequency would be possible if measurements were known with different wavelengths, which is not the case. Therefore, the periodic calculation can be used as benchmark for the efficiency of the other theoretical approaches. In this respect, we observe that RLFT substantially improves over the oriented-gas, but still underperforms the periodic first-principles calculation. Interestingly, the electric susceptibility tensor and refractive indices of crystalline urea have been estimated from a variety of approaches [
13]. Among them, RLFT resulted in values very close to the periodic first-principles methods, as well as to the experimental values extrapolated to zero frequency. The application of RLFT to a urea molecule extracted from a relatively large cluster slightly overcorrects the refractive indices.
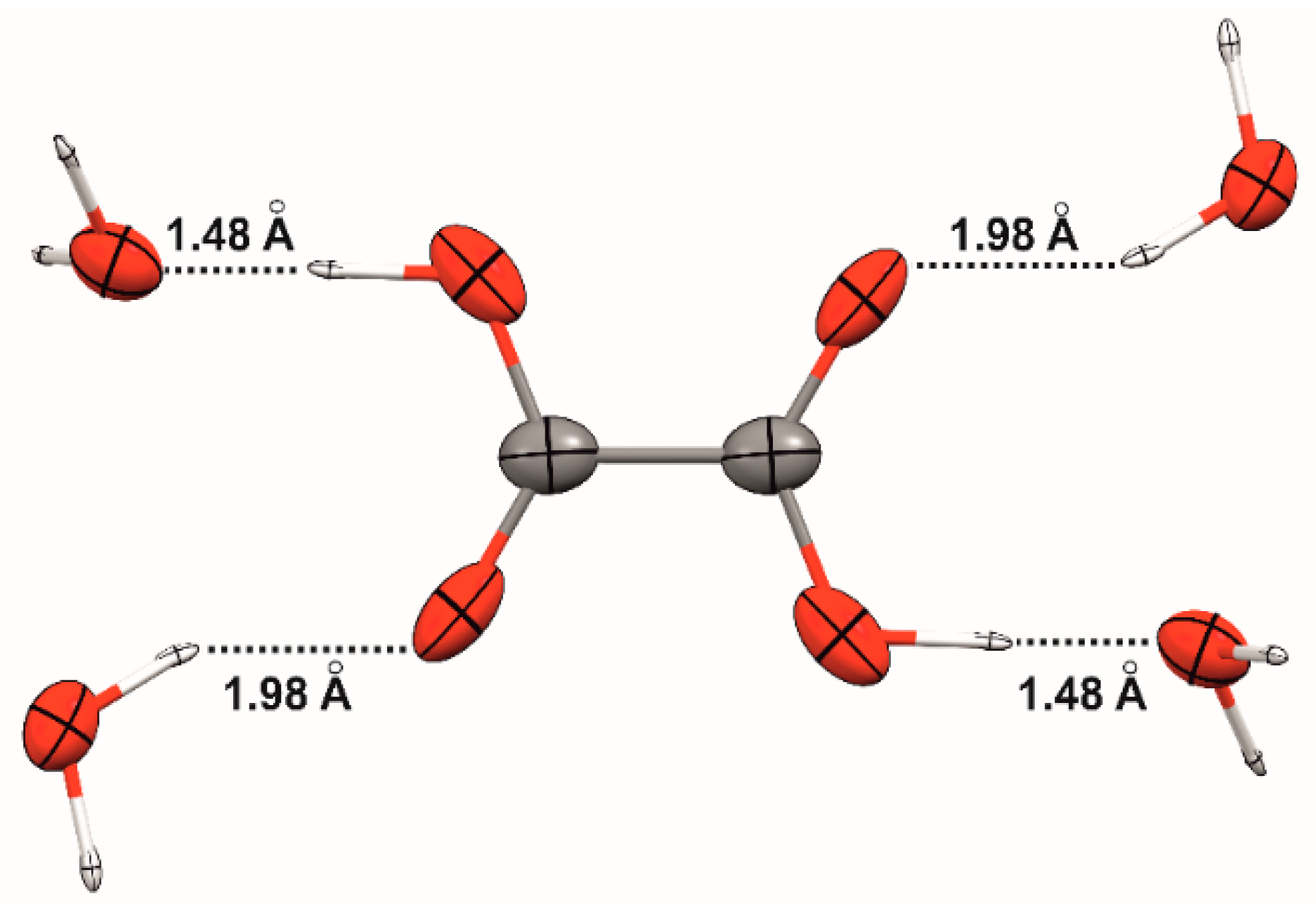
Finally, we consider the example of three aggregates composed of oxalic acid and water molecules (see
Table 3 and
Figure 5). Overall, the polarizability of the carboxylate groups are not dramatically affected by the hydrogen bonds formed with the solvent molecules, even though such interactions may be considered rather strong. Indeed, the –COOH isotropic polarizability increases only 4% when going from the oxalic acid in isolation to the tetra-hydrate cluster. The enhancement is more significant along the direction of the hydrogen-bond axes. Interestingly, the shorter hydrogen bonds (where oxalic acid acts as a donor) change less the oxalic acid polarizability than the longer ones (where oxalic acid is the acceptor). In this respect, it should also be taken into account that the volume associated with the –COOH group in the C
2H
2O
4·2 H
2O (sHB) aggregate is inherently smaller than that in the C
2H
2O
4·2 H
2O (mHB) aggregate (sHB is the aggregate produced by the shorter HB, mHB is the medium length HB adduct). Therefore, the fact that the
tensor component of the –COOH polarizability in sHB is smaller than that in mHB is a manifestation not only of the extra electron-density polarization caused by the presence of the water molecules, but also of the change in volume undergone by the oxalic acid when hydrated, with respect to the anhydrous form. The
ratio for the –COOH group (with
calculated using a 0.001 au isodensity surface) is in fact constant for the whole series of aggregates.









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}