
Results from Exploratory Work in Li-Rich Regions of the AE-Li-Ge Systems (AE = Ca, Sr, Ba)

Abstract
:1. Introduction
2. Materials and Methods
2.1. Synthesis
2.2. Crystallographic Analysis
2.3. Elemental Analysis
2.4. Electronic Structure Calculations
2.5. Electrochemical Measurements
3. Results and Discussion
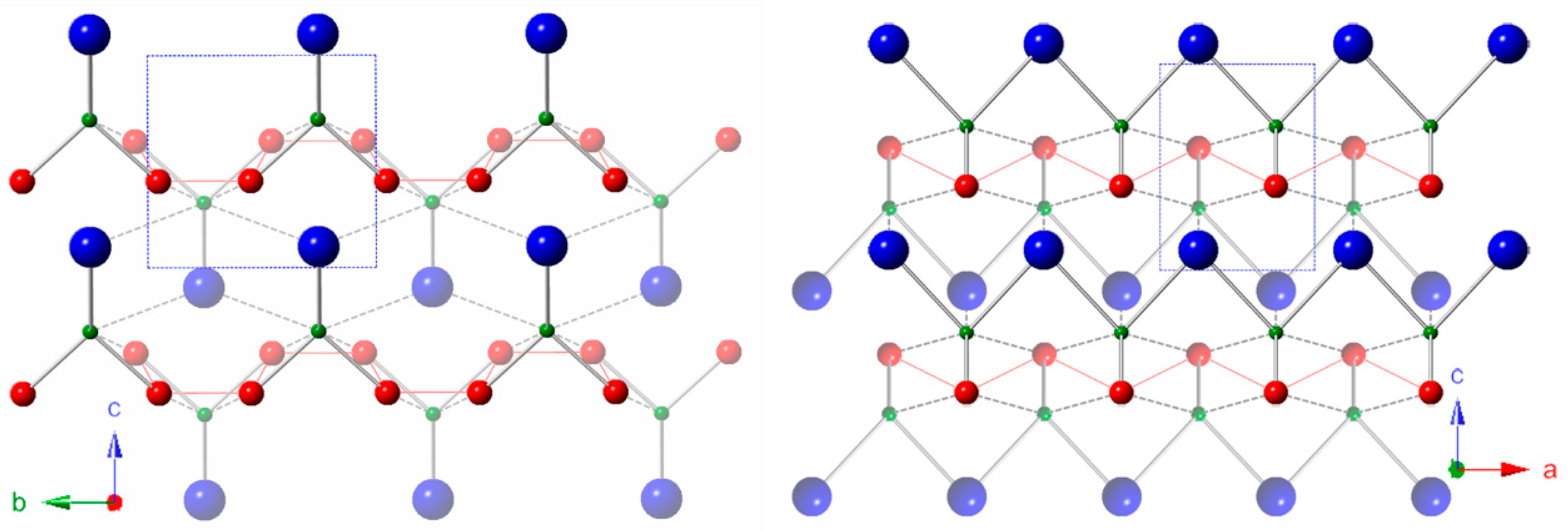
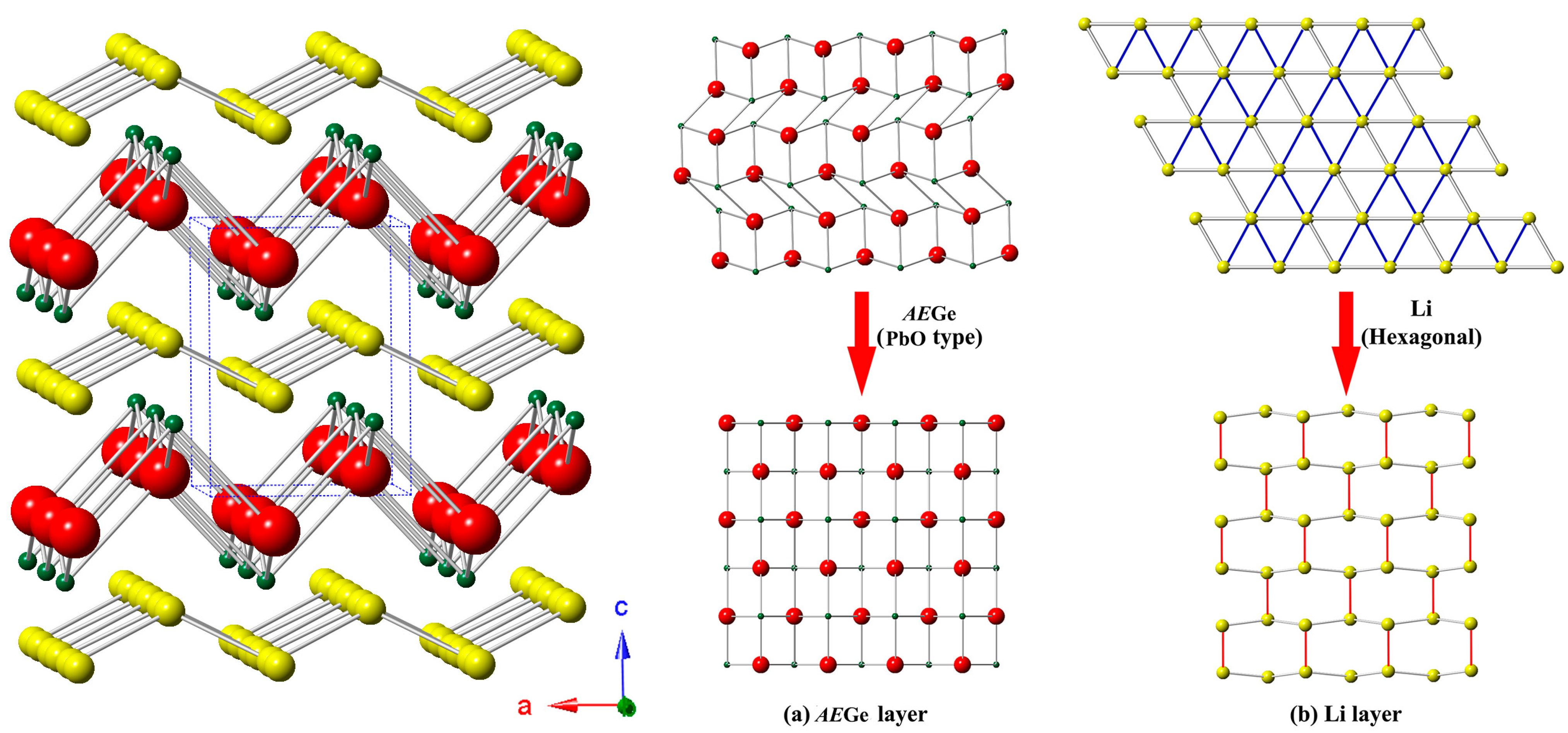
3.1. Crystal Structure
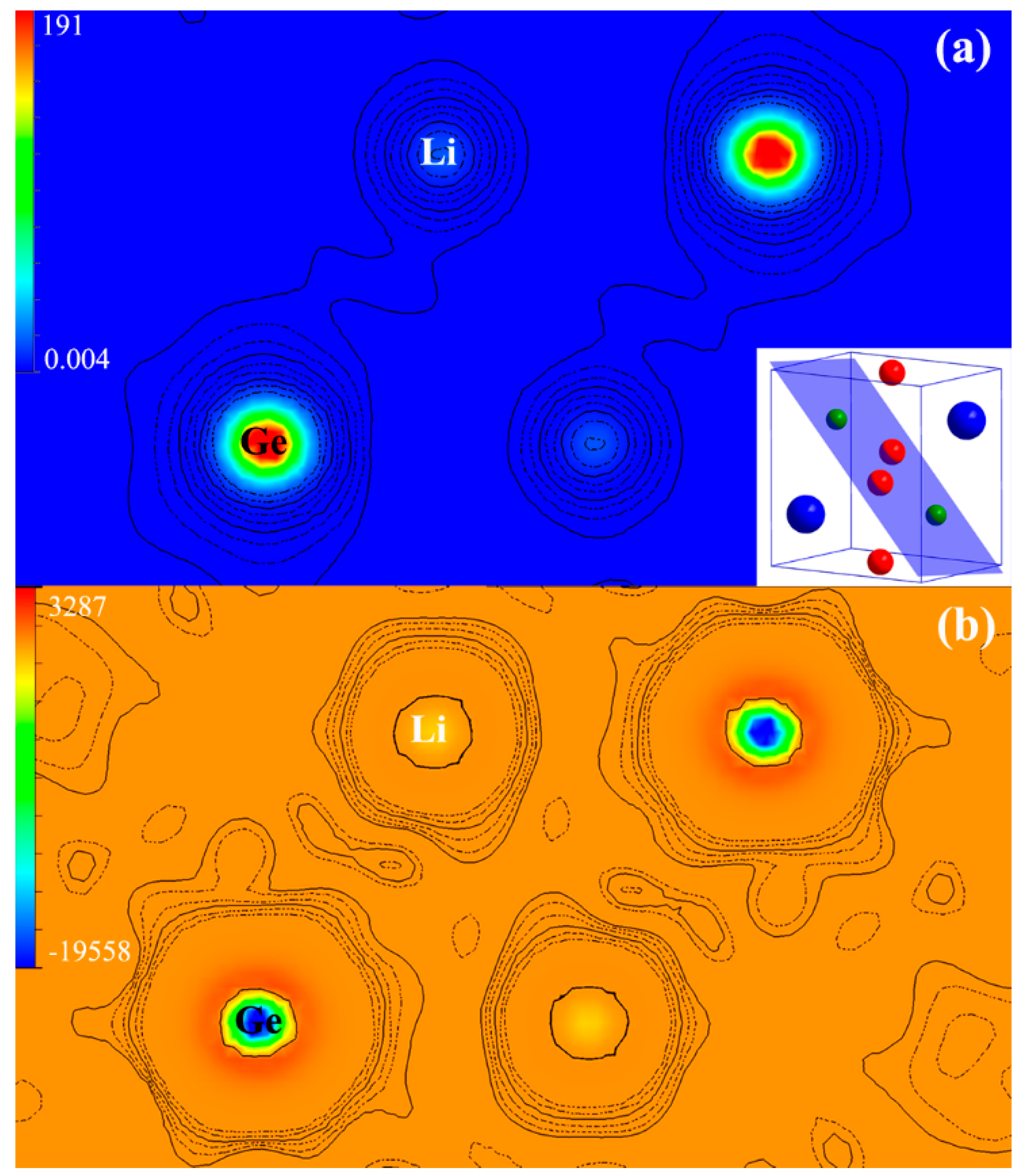
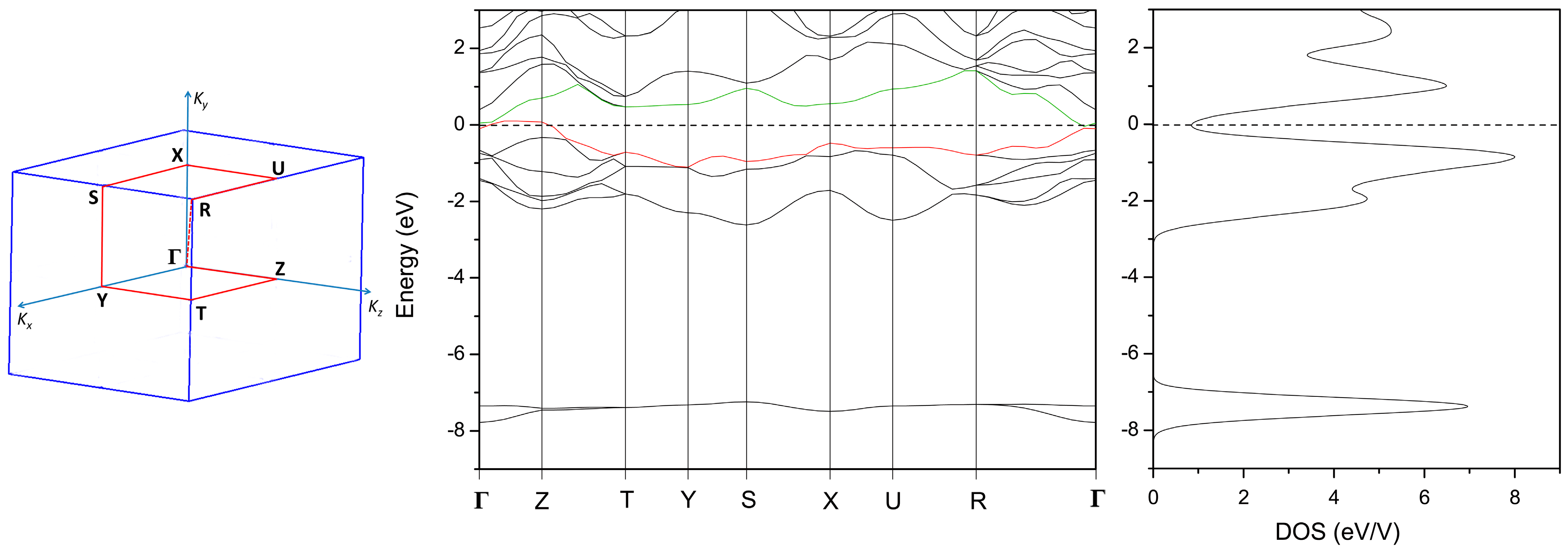
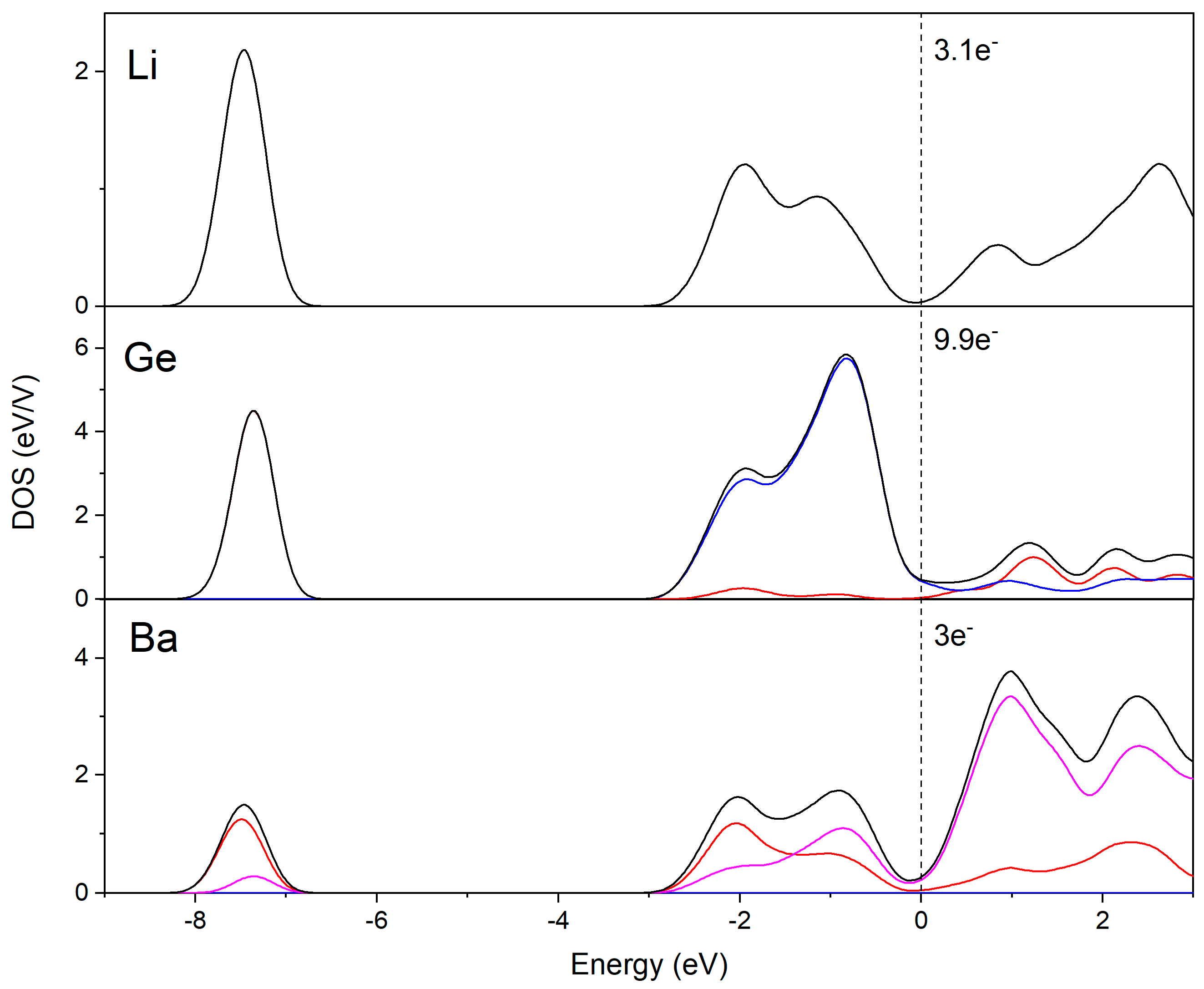
3.2. Electronic Structure
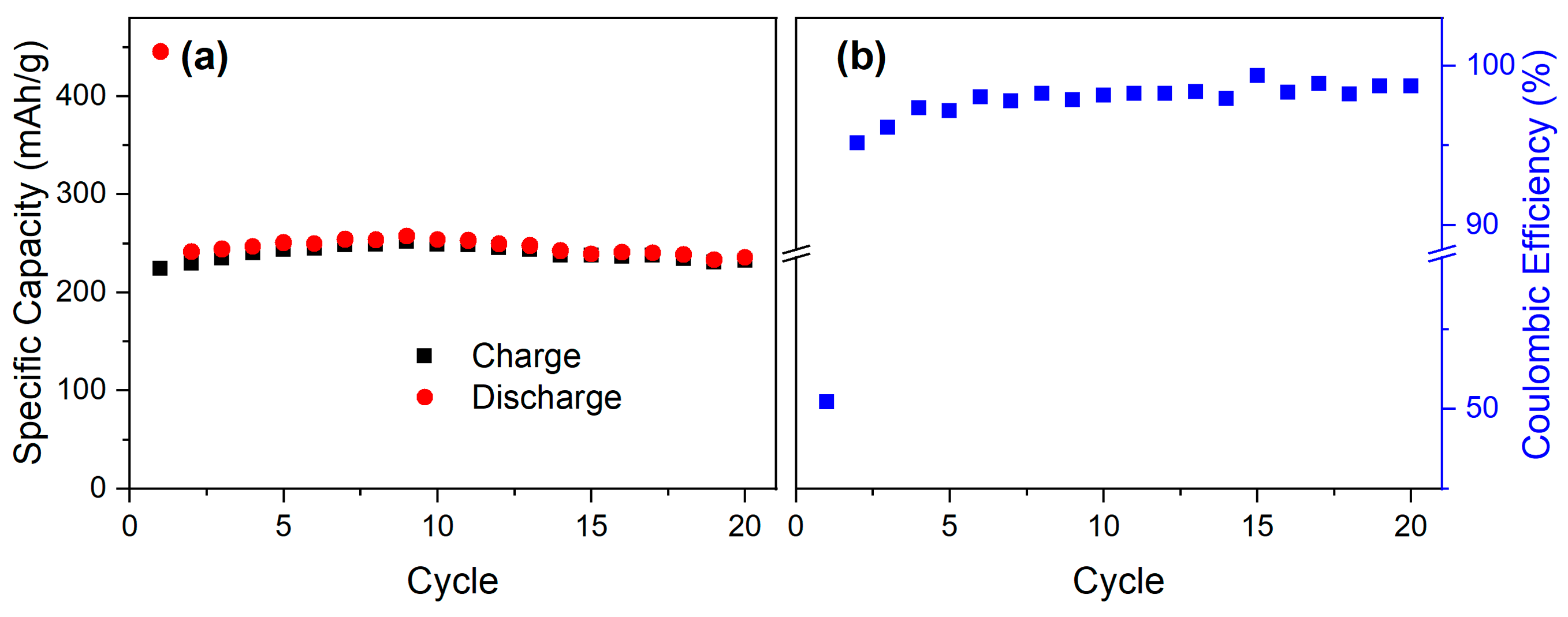
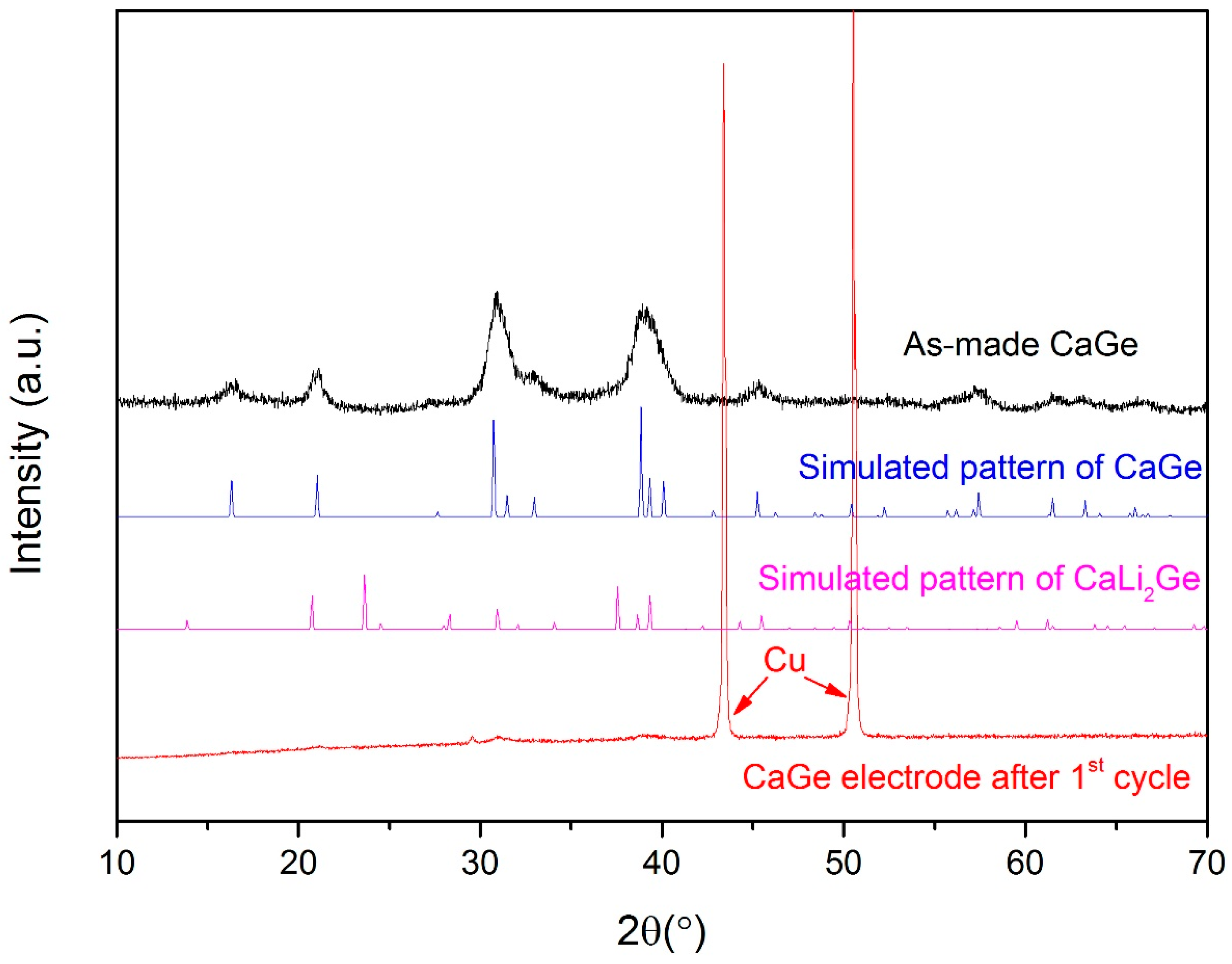
3.3. Electrochemical Properties
4. Conclusions
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
Appendix A

References
- Whittingham, M.S. Lithium Batteries and Cathode Materials. Chem. Rev. 2004, 104, 4271–4301. [Google Scholar] [CrossRef] [PubMed]
- Goodenough, J.B.; Park, K.S. The Li-Ion Rechargeable Battery: A Perspective. J. Am. Chem. Soc. 2013, 135, 1167–1176. [Google Scholar] [CrossRef] [PubMed]
- Goodenough, J.B.; Kim, Y. Challenges for Rechargeable Li Batteries. Chem. Mater. 2010, 22, 587–603. [Google Scholar] [CrossRef]
- Sathiya, M.; Rousse, G.; Ramesha, K.; Laisa, C.P.; Vezin, H.; Sougrati, M.T.; Doublet, M.L.; Foix, D.; Gonbeau, D.; Walker, W.; et al. Reversible Anionic Redox Chemistry in High-Capacity Layered-Oxide Electrodes. Nat. Mater. 2013, 12, 827–835. [Google Scholar] [CrossRef]
- Goodenough, J.B. Evolution of Strategies for Modern Rechargeable Batteries. Acc. Chem. Res. 2013, 46, 1053–1061. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.; Urban, A.; Li, X.; Su, D.; Hautier, G.; Ceder, G. Unlocking the potential of cation-disordered oxides for rechargeable lithium batteries. Science 2014, 343, 519. [Google Scholar] [CrossRef]
- Collins, C.; Dyer, M.S.; Pitcher, M.J.; Whitehead, G.F.S.; Zanella, M.; Mandal, P.; Claridge, J.B.; Darling, G.R.; Rosseinsky, M.J. Accelerated Discovery of Two Crystal Structure Types in a Complex Inorganic Phase Field. Nature 2017, 546, 280. [Google Scholar] [CrossRef]
- Dyer, M.S.; Collins, C.; Hodgeman, D.; Chater, P.A.; Demont, A.; Romani, S.; Sayer, R.; Thomas, M.F.; Claridge, J.B.; Darling, G.R.; et al. Computationally Assisted Identification of Functional Inorganic Materials. Science 2013, 340, 847–852. [Google Scholar] [CrossRef]
- Longo, R.C.; Kong, F.T.; Santosh, K.C.; Park, M.S.; Yoon, J.; Yeon, D.H.; Park, J.H.; Doo, S.G.; Cho, K. Phase Stability of Li-Mn-O Oxides as Cathode Materials for Li-ion Batteries: Insights from Ab Initio Calculation. Phys. Chem. Chem. Phys. 2014, 16, 11218. [Google Scholar] [CrossRef]
- Matsuno, S.; Noji, M.; Kashiwagi, T.; Nakayama, M.; Wakihara, M. Construction of the Ternary Phase Diagram for the Li-Cu-Sb System as the Anode Materials for a Lithium Ion Battery. J. Phys. Chem. C 2007, 111, 7548–7553. [Google Scholar] [CrossRef]
- Massalski, T.B. (Ed.) Binary Alloys Phase Diagrams; ASM International: Materials Park, OH, USA, 1990. [Google Scholar]
- St John, M.R.; Furgala, A.J.; Sammells, A.F. Thermodynamic Studies of Li-Ge Alloys: Application to Negative Electrodes for Molten Salt Batteries. J. Electrochem. Soc. 1982, 129, 246–250. [Google Scholar] [CrossRef]
- Vhof, V.; Muller, W.; Schafer, H. The Structure of Li7Ge2. Z. Naturforsch. B 1972, 27B, 1157–1160. [Google Scholar]
- Fässler, T.F.; Zeilinger, M. Structural and Thermodynamic Similarities of Phases in the Li-Tt (Tt = Si, Ge) Systems: Redetermination of the Lithium-rich Side of the Li-Ge Phase Diagram and Crystal Structure of Li17Si4−xGex for x = 2.3, 3.1, 3.5 and 4 As Well As Li4.1Ge. Dalton Trans. 2014, 43, 14959–14970. [Google Scholar]
- Johnson, Q.; Smith, G.S.; Wood, D. The Crystal Structure of Li15Ge4. Acta Crystallogr. 1965, 18, 131–132. [Google Scholar] [CrossRef]
- Osman, H.H.; Bobev, S. Experimental and Theoretical Study on the Substitution Patterns in Lithium Germanides: The Case of Li15Ge4 vs. Li14ZnGe4. Eur. J. Inorg. Chem. 2022, 2022, e202100901. [Google Scholar] [CrossRef]
- Goward, G.R.; Taylor, N.J.; Souza, D.C.S.; Nazar, L. The True Crystal Structure of Li17M4 (M = Ge, Sn, Pb)—Revised from Li22M5. J. Alloys Compd. 2001, 329, 82–91. [Google Scholar] [CrossRef]
- Tillard, M.; Belin, C.; Spina, L.; Jia, Y.Z. Two Cubic Polymorphs of AlGeLi. Acta Crystallogr. Sect. C 2012, 68, i60–i64. [Google Scholar]
- Henze, A.; Hlukhyy, V.; Fässler, T.F. Fully and Partially Li-Stuffed Diamond Polytypes with Ag-Ge Structures: Li2AgGe and Li2.53AgGe2. Inorg. Chem. 2015, 54, 1152–1158. [Google Scholar] [CrossRef]
- Todorov, I.; Sevov, S.C. Heavy-Metal Aromatic and Conjugated Species: Rings, Oligomers, and Chains of Tin in Li9−xEuSn6+x, Li9−xCaSn6+x, Li5Ca7Sn11, Li6Eu5Sn9, LiMgEu2Sn3, and LiMgSr2Sn3. Inorg. Chem. 2005, 44, 5361–5369. [Google Scholar] [CrossRef]
- Ganguli, A.K.; Gupta, S.; Corbett, J.D. New Tetragonal Structure Type for A2Ca10Sb9 (A = Li, Mg). Electronic Variability Around a Zintl Phase. Inorg. Chem. 2006, 45, 196–200. [Google Scholar] [CrossRef]
- Stegmaier, S.; Waibel, M.; Henze, A.; Jantke, L.A.; Karttunen, A.J.; Fässler, T.F. Soluble Zintl phase A14ZnGe16 (A = K, Rb) Featuring [(η3-Ge4)Zn(η3-Ge4)]6− and [Ge4]4− Clusters and the Isolation of [(MesCu)2(η3-Ge4)Zn(η3,η3-Ge4)]4−: The Missing Link in the Solution Chemistry of Tetrahedral Group 14 Element Zintl Clusters. J. Am. Chem. Soc. 2012, 134, 14450–14460. [Google Scholar] [PubMed]
- Luo, X.H.; Krizan, J.W.; Haldolaarachchige, N.; Klimczuk, T.; Xiw, W.W.; Fuccillo, M.K.; Felser, C.; Cava, R.J. A Large Family of Filled Skutterudites Stabilized by Electron Count. Nat. Commun. 2015, 6, 6489. [Google Scholar] [PubMed]
- Schäfer, M.C.; Bobev, S. Tin Clathrates with the Type II Structure. J. Am. Chem. Soc. 2013, 135, 1696. [Google Scholar] [CrossRef] [PubMed]
- Fässler, T.F. (Ed.) Zintl Ions: Principles and Recent Developments; Springer: Berlin/Heidelberg, Germany, 2011. [Google Scholar]
- Stoiber, D.; Bobnar, M.; Höhn, P.; Niewa, R. Lithium Alkaline Earth Tetrelides of the type Li2AeTt (Ae = Ca, Ba, Tt = Si, Ge, Sn, Pb): Synthesis, Crystal Structures and Physical Properties. Z. Naturforsch. B 2017, 72, 220–222. [Google Scholar] [CrossRef]
- SMART NT, Version 5.63; Bruker Analytical X-ray Systems: Madison, WI, USA, 2003.
- SAINT NT, Version 6.45; Bruker Analytical X-ray Systems: Madison, WI, USA, 2003.
- SADABS NT, Version 2.10; Bruker Analytical X-ray Systems: Madison, WI, USA, 2001.
- SHELXTL, Version 6.12; Bruker Analytical X-ray Systems: Madison, WI, USA, 2001.
- Momma, K.; Ikeda, T.; Belik, A.A.; Izumi, F. Dysnomia, A Computer Program for Maximum-Entropy Method (MEM) Analysis and its Performance in the MEM-Based Pattern Fitting. Powder Diffr. 2013, 28, 184–193. [Google Scholar] [CrossRef]
- Momma, K.; Izumi, F. VESTA 3 for Three-Dimensional Visualization of Crystal, Volumetric and Morphology Data. J. Appl. Cryst. 2011, 44, 1272–1276. [Google Scholar] [CrossRef]
- Ordejόn, P.; Artacho, E.; Soler, J.M. Self-consistent Order-N Density-Functional Calculations for Very Large Systems. Phys. Rev. B 1996, 53, R10441. [Google Scholar] [CrossRef]
- Soler, J.M.; Artacho, E.; Gale, J.D.; García, A.; Junquera, J.; Ordejόn, P.; Sánchez-Portal, D. The SIESTA Method for Ab Initio Order-N Materials Simulation. J. Phys. Condens. Matter 2002, 14, 2745. [Google Scholar] [CrossRef]
- Troullier, N.; Martins, J.L. Efficient Pseudopotentials for Plane-Wave Calculations. Phys. Rev. B 1991, 43, 1993. [Google Scholar] [CrossRef]
- Perdew, J.P.; Burke, K.; Emzerhof, M. Generalized Gradient Approximation Made Simple. Phys. Rev. Lett. 1996, 77, 3865. [Google Scholar] [CrossRef]
- Kresse, G.; Hafner, J. Ab Initio Molecular Dynamics for Liquid Metals. Phy. Rev. B 1993, 47, 558. [Google Scholar] [CrossRef] [PubMed]
- Kresse, G.; Joubert, D. From Ultrasoft Pseudopotential to the Projector Augmented-Wave Method. Phys. Rev. B 1999, 59, 1758. [Google Scholar] [CrossRef]
- Von Narth, U.; Hedin, L.A. A Local Exchange-Correlation Potential for the Spin Polarized Case. I. J. Phys. C 1972, 5, 1629. [Google Scholar]
- Betz, A.; Schafer, H.; Weiss, A.; Wulf, R. Digermanide SrGe2 and BaGe2. Z. Naturforsch. B 1968, 23, 878. [Google Scholar] [CrossRef]
- Park, D.G.; Dong, Y.; DiSalvo, F.J. LiSrGe2 and LiBaGe2: One-Dimensional Chains of [Ge2]3− in an Unusual Conformation. J. Alloys Compd. 2009, 470, 90–95. [Google Scholar] [CrossRef]
- von Schnering, H.G.; Bolle, U.; Curde, J.; Peter, K.; Carillo-Cabrera, W.; Mehmet, S.; Schultheiss, M.; Wedig, U. Hucker Arenes with Ten π Electrons: Cyclic Zintl Anions [Si6]10− and [Ge6]10−, Isosteric with [P6]4−. Angew. Chem. 1996, 35, 984–986. [Google Scholar] [CrossRef]
- Ghosh, K.; Bobev, S. Yet Another Case of Lithium Metal Atoms and Germanium Atoms Sharing Chemistry in the Solid State: Synthesis and Structural Characterization of Ba2LiGe3. Chem. Eur. J. 2023, 29, e202302385. [Google Scholar] [CrossRef]
- Ghosh, K.; Ovchinnikov, A.; Baitinger, M.; Krnel, M.; Burkhardt, U.; Grin, Y.; Bobev, S. Lithium Metal Atoms Fill Vacancies in the Germanium Network of a Type-I Clathrate: Synthesis and Structural Characterization of Ba8Li5Ge41. Dalton Trans. 2023, 52, 10310. [Google Scholar] [CrossRef]
- Guo, S.P.; You, T.S.; Bobev, S. Closely Related Rare-Earth Metal Germanides RE2Li2Ge3 and RE3Li4Ge4 (RE = La-Nd, Sm): Synthesis, Crystal Chemistry, and Magnetic Properties. Inorg. Chem. 2012, 51, 3119–3129. [Google Scholar] [CrossRef]
- Lacroix-Orio, L.; Tillard, M.; Belin, C. Synthesis, Crystal and Electronic Structure of Li8Zn2Ge3, a Compound Displaying an Open Layered Anionic Network. Solid State Sci. 2006, 8, 208–215. [Google Scholar] [CrossRef]
- Howard, W.F.; Spotnitz, R.M. Theoretical Evaluation of High-Energy Lithium Metal Phosphate Cathode Materials in Li-ion Batteries. J. Power Source 2007, 165, 887. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| CCDC number | 2314734 | 2314735 | 2314736 |
| Empirical formula | CaLi2Ge | SrLi2Ge | BaLi2Ge |
| Formula weight | 126.55 | 174.09 | 223.81 |
| Temperature | 200(2) K | ||
| Radiation, λ | Mo Kα, 0.71073 Å | ||
| Space group | Pmmn (no. 59), Z = 2 | ||
| Lattice constants | |||
| a/Å | 4.650 (2) | 4.6818 (7) | 4.7092 (9) |
| b/Å | 5.766 (3) | 6.2829 (6) | 6.7593 (9) |
| c/Å | 6.364 (3) | 6.2800 (7) | 6.2923 (7) |
| V/Å 3 | 170.65 (13) | 184.81 (4) | 200.3 (1) |
| ρ/g·cm−3 | 2.46 | 3.13 | 3.60 |
| μ/cm−1 | 101.7 | 222.8 | 170.1 |
| R1 (I > 2σ(I)) 1 | 0.0266 | 0.009 | 0.0104 |
| wR2 (I > 2σ(I)) 1 | 0.0544 | 0.0209 | 0.0201 |
| R1 (all data) 1 | 0.0332 | 0.0094 | 0.0114 |
| wR2 (all data) 1 | 0.0566 | 0.021 | 0.0268 |
| Largest diff. peak/hole | 0.55/–0.67 | 0.38/–0.32 | 0.41/–0.51 |
| Atom | Site | x | y | z | Ueq (Å) |
|---|---|---|---|---|---|
| CaLi2Ge | |||||
| Ca | 2b | 1/4 | 1/4 | 0.0678(3) | 0.0135(4) |
| Ge | 2b | 1/4 | 3/4 | 0.2582(2) | 0.0123(3) |
| Li | 4e | 0 | 0.0251(15) | 0.6018(15) | 0.023(2) |
| SrLi2Ge | |||||
| Sr | 2b | 1/4 | 1/4 | 0.0885(1) | 0.0108(2) |
| Ge | 2b | 1/4 | 3/4 | 0.2793(1) | 0.0102(2) |
| Li | 4e | 0 | 0.0369(7) | 0.5991(6) | 0.0169(8) |
| BaLi2Ge | |||||
| Ba | 2b | 1/4 | 1/4 | 0.0981(1) | 0.0126(2) |
| Ge | 2b | 1/4 | 3/4 | 0.3027(2) | 0.0110(2) |
| Li | 4e | 0 | 0.0483(8) | 0.5935(8) | 0.016(1) |
| Atomic Pair | Distance (Å) | Atomic Pair | Distance (Å) | Atomic Pair | Distance (Å) |
|---|---|---|---|---|---|
| CaLi2Ge | SrLi2Ge | BaLi2Ge | |||
| Ge—Ca (×2) | 3.117(2) | Ge—Sr (×2) | 3.2891(4) | Ge—Ba (×2) | 3.4503(4) |
| Ge—Ca (×2) | 3.127(2) | Ge—Sr (×2) | 3.3623(3) | Ge—Ba (×2) | 3.6166(4) |
| Ge—Li (×2) | 2.701(9) | Ge—Li (×2) | 2.700(4) | Ge—Li (×2) | 2.7228(2) |
| Ge—Li (×4) | 2.808(2) | Ge—Li (×4) | 2.803(2) | Ge—Li (×2) | 2.7981(2) |
| Li—Li (×1) | 2.59(1) | Li—Li (×1) | 2.677(8) | Li—Li (×2) | 2.7120(2) |
| Li—Li (×2) | 2.678(9) | Li—Li (×2) | 2.692(3) | Li—Li (×1) | 3.7267(4) |
| Ca—Li (×2) | 3.237(9) | Sr—Li (×2) | 3.353(4) | Ba—Li (×2) | 3.4023(3) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, J.; Bobev, S. Results from Exploratory Work in Li-Rich Regions of the AE-Li-Ge Systems (AE = Ca, Sr, Ba). Crystals 2024, 14, 57. https://doi.org/10.3390/cryst14010057
Zhang J, Bobev S. Results from Exploratory Work in Li-Rich Regions of the AE-Li-Ge Systems (AE = Ca, Sr, Ba). Crystals. 2024; 14(1):57. https://doi.org/10.3390/cryst14010057
Chicago/Turabian StyleZhang, Jiliang, and Svilen Bobev. 2024. "Results from Exploratory Work in Li-Rich Regions of the AE-Li-Ge Systems (AE = Ca, Sr, Ba)" Crystals 14, no. 1: 57. https://doi.org/10.3390/cryst14010057