
Stability and Elasticity of Quasi-Hexagonal Fullerene Monolayer from First-Principles Study
 , ,
, , 
Abstract
:1. Introduction
2. Materials and Methods
3. Results
3.1. Crystal Structure, Thermal, and Structural Stability
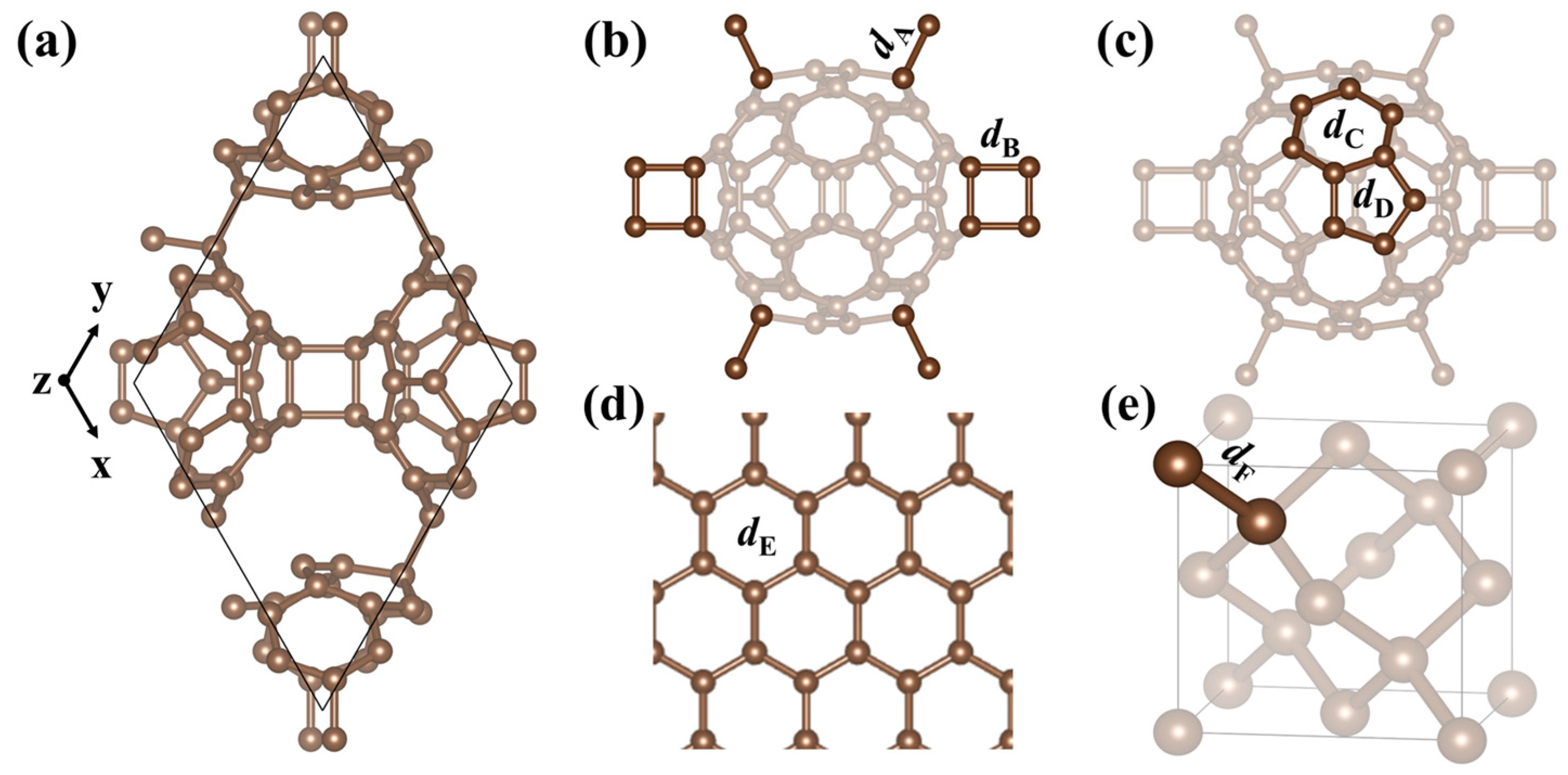
3.1.1. Crystal Structure
3.1.2. Formation and Binding Energy
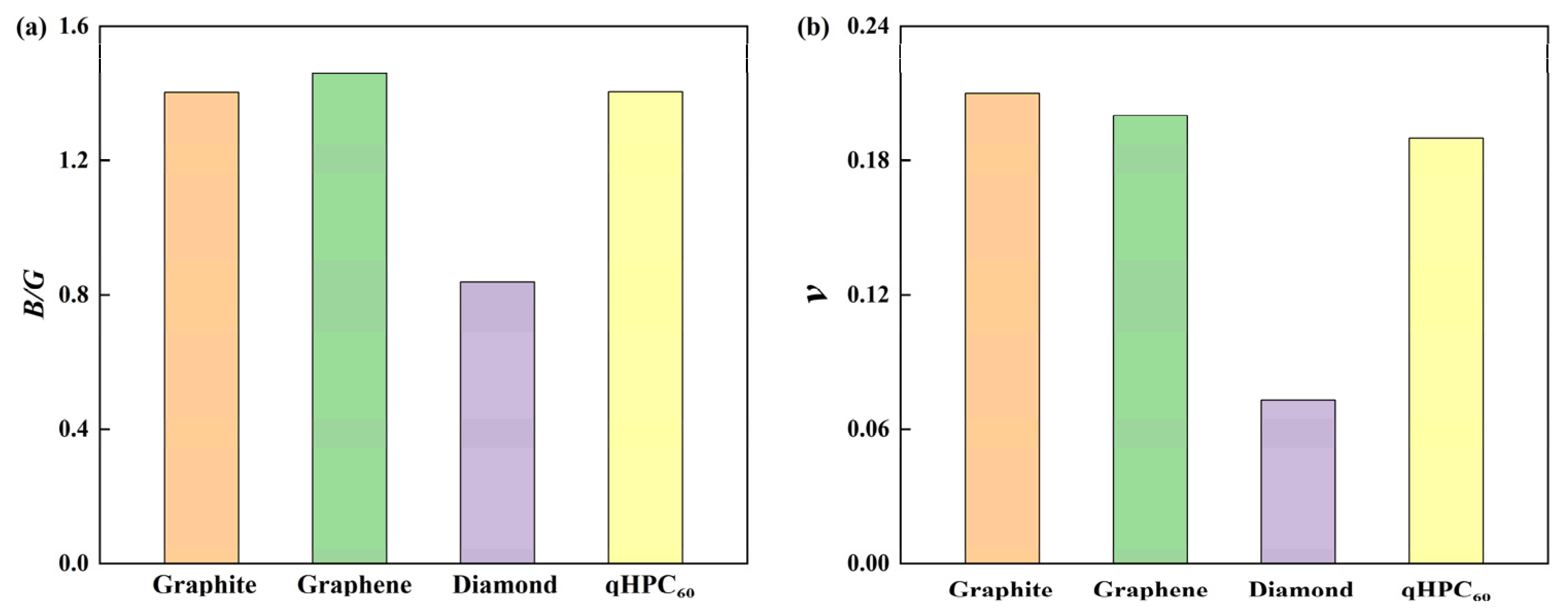
3.2. Mechanical Stability
3.2.1. Elasticity Constants
3.2.2. Ductility
3.2.3. Hardness
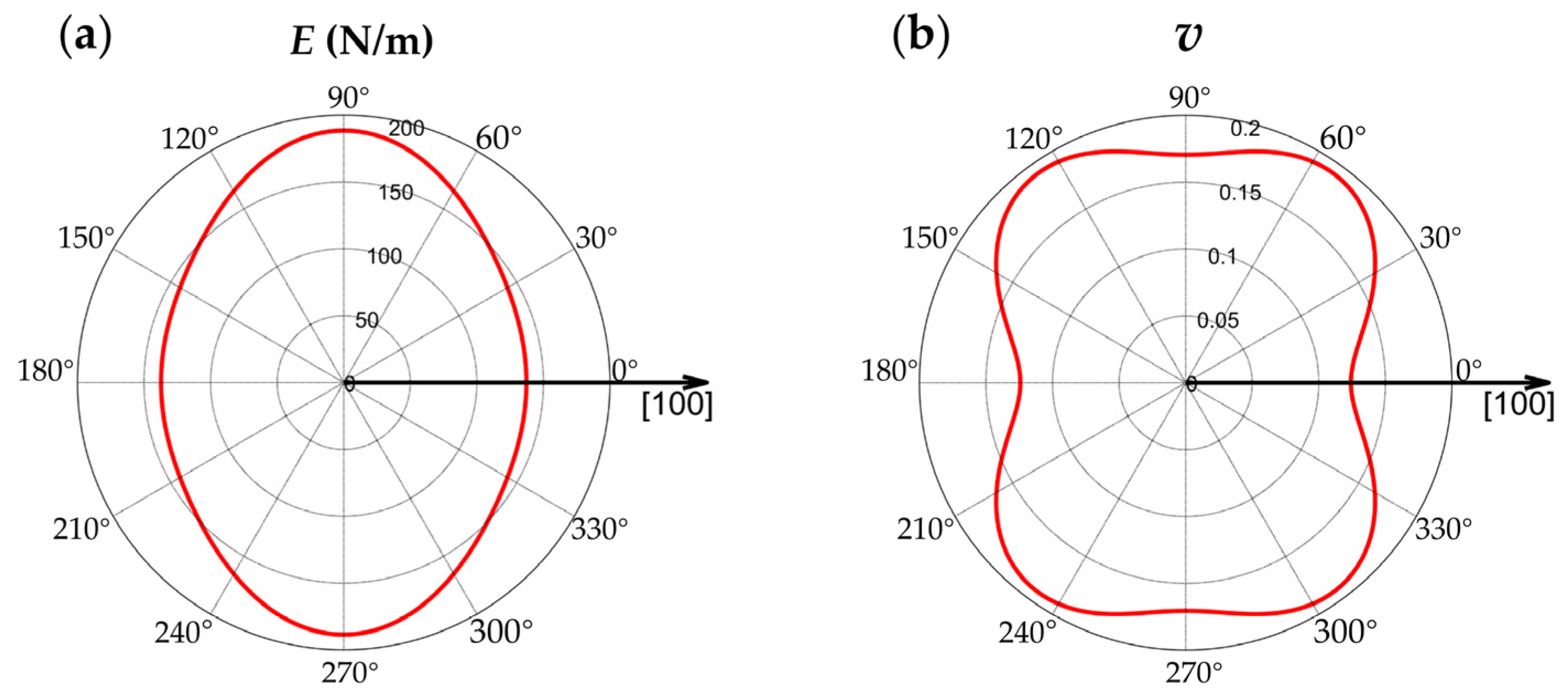
3.2.4. Anisotropy of the Material
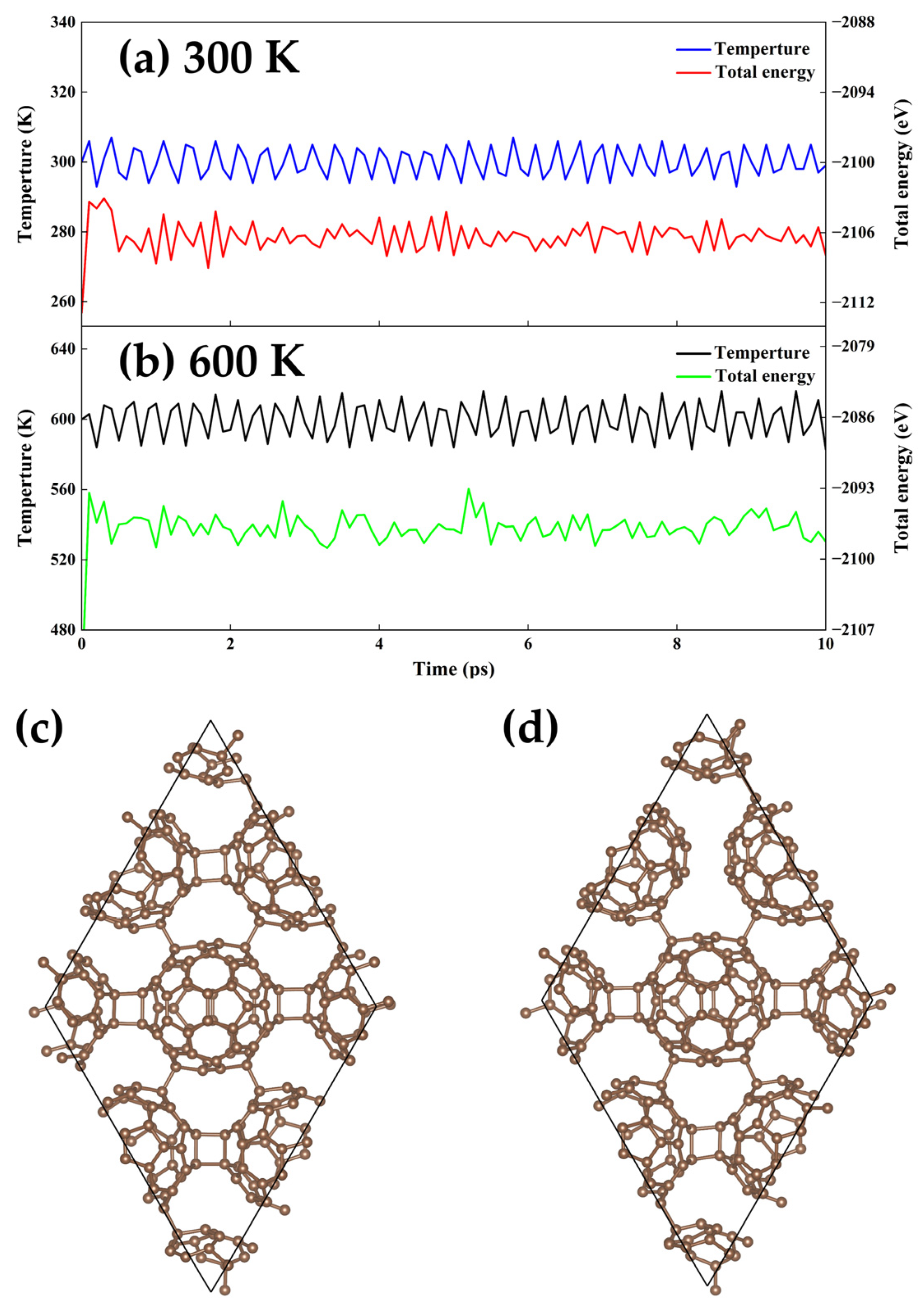
3.3. Thermodynamic Stability
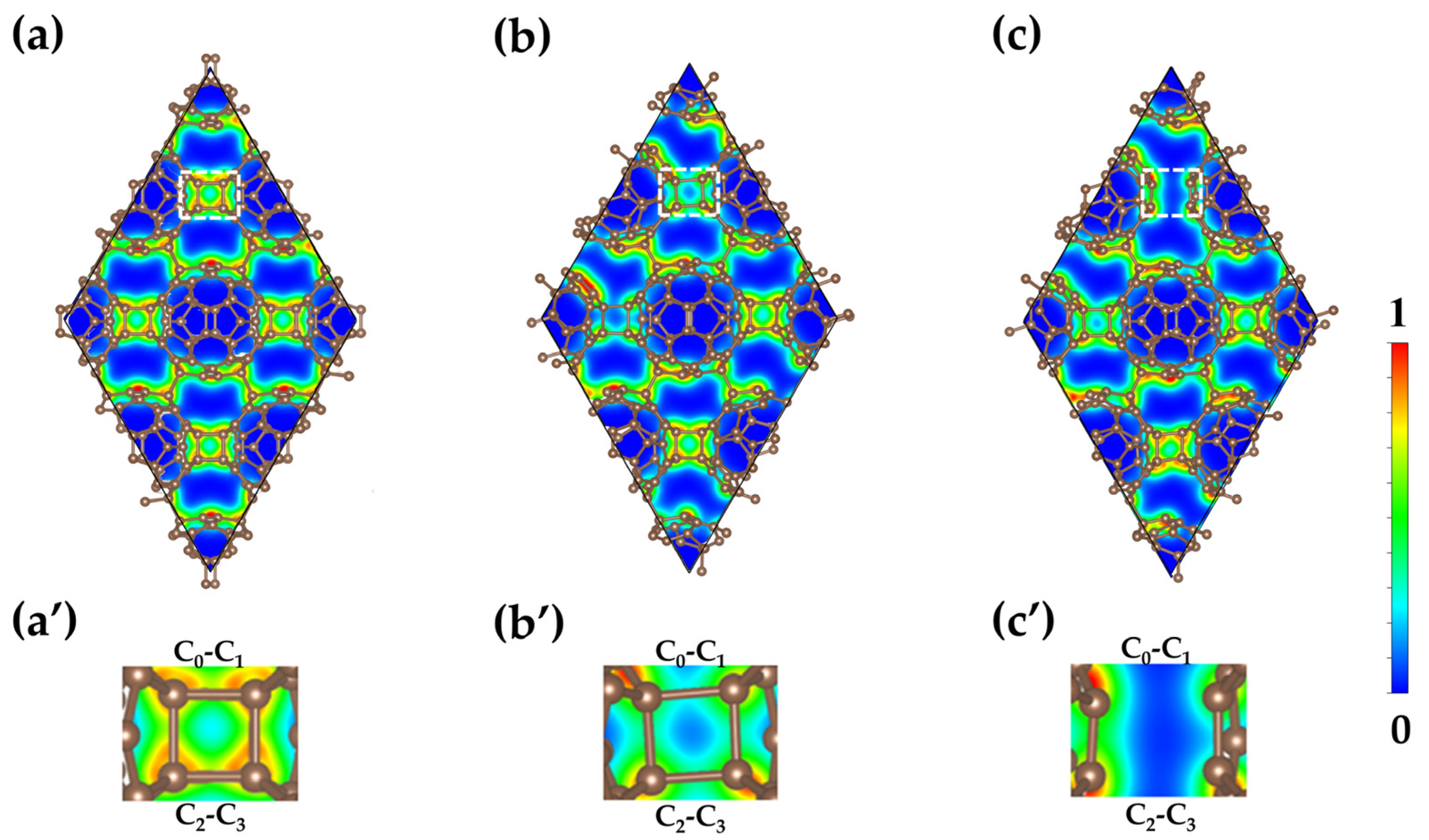
3.4. Electronic Localization Function Analysis
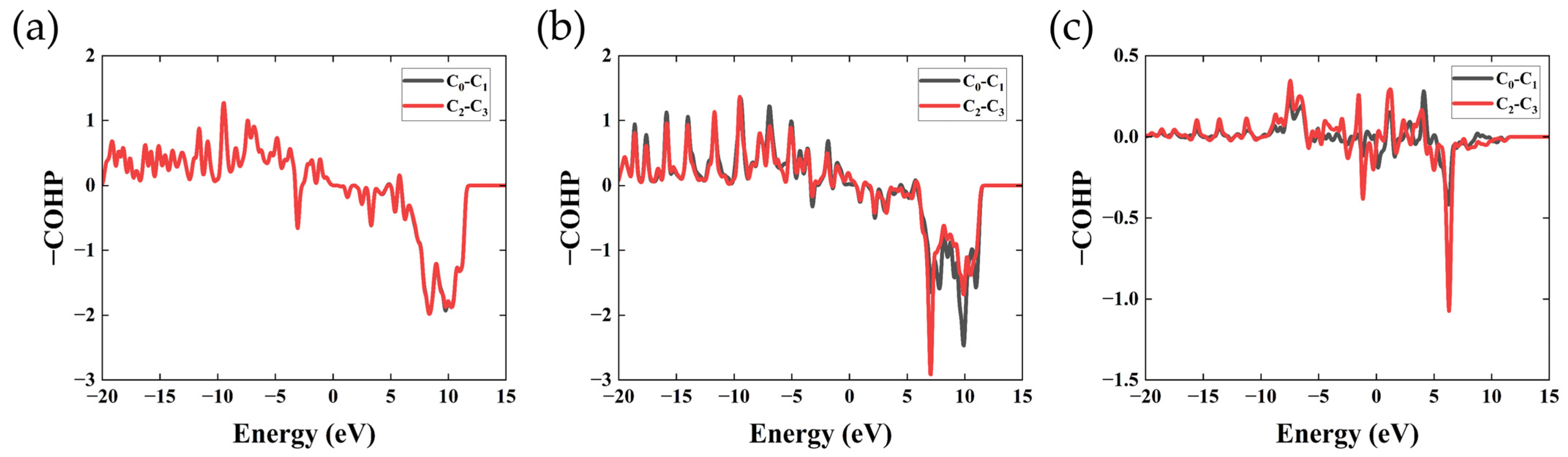
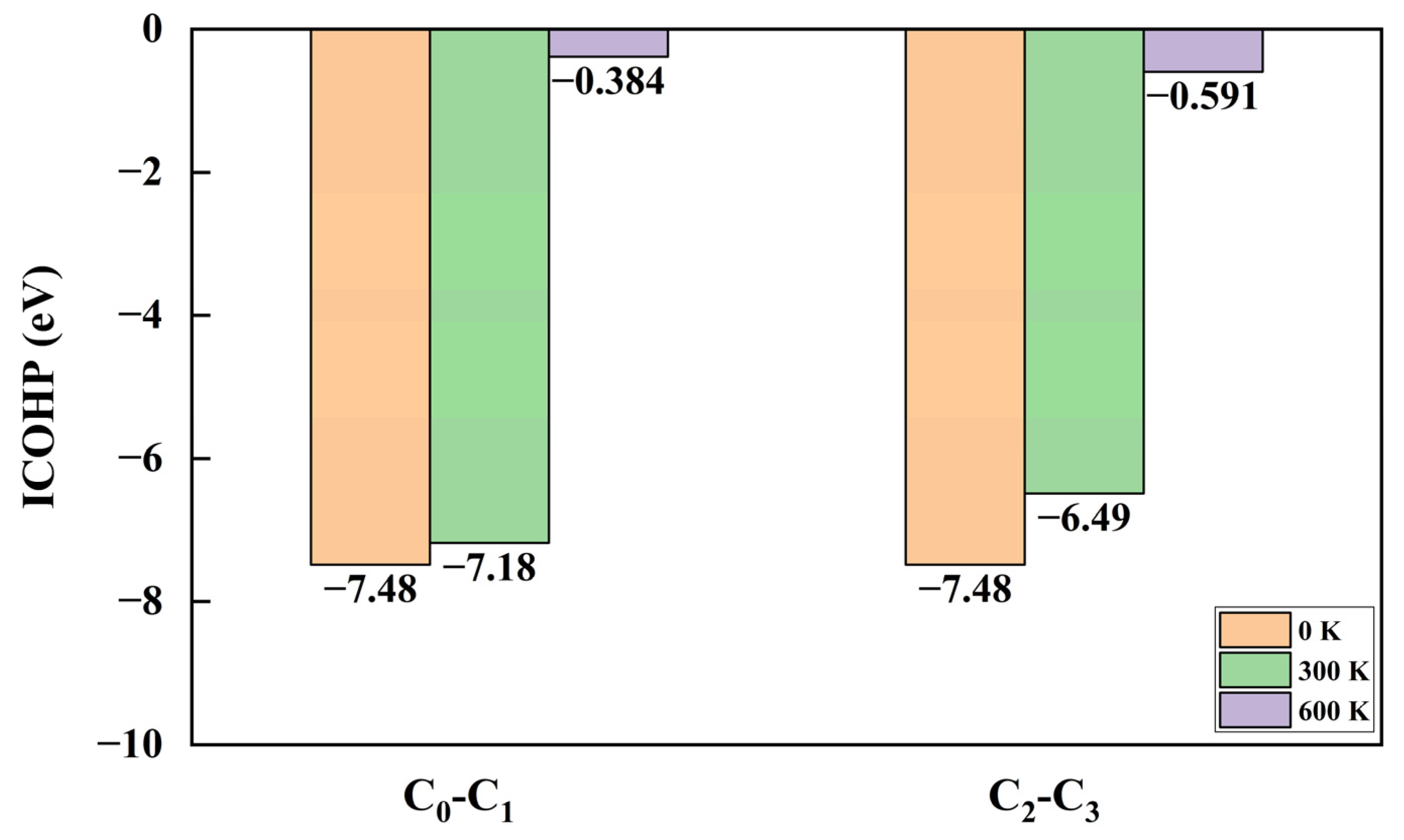
3.5. COHP and ICOHP Analysis
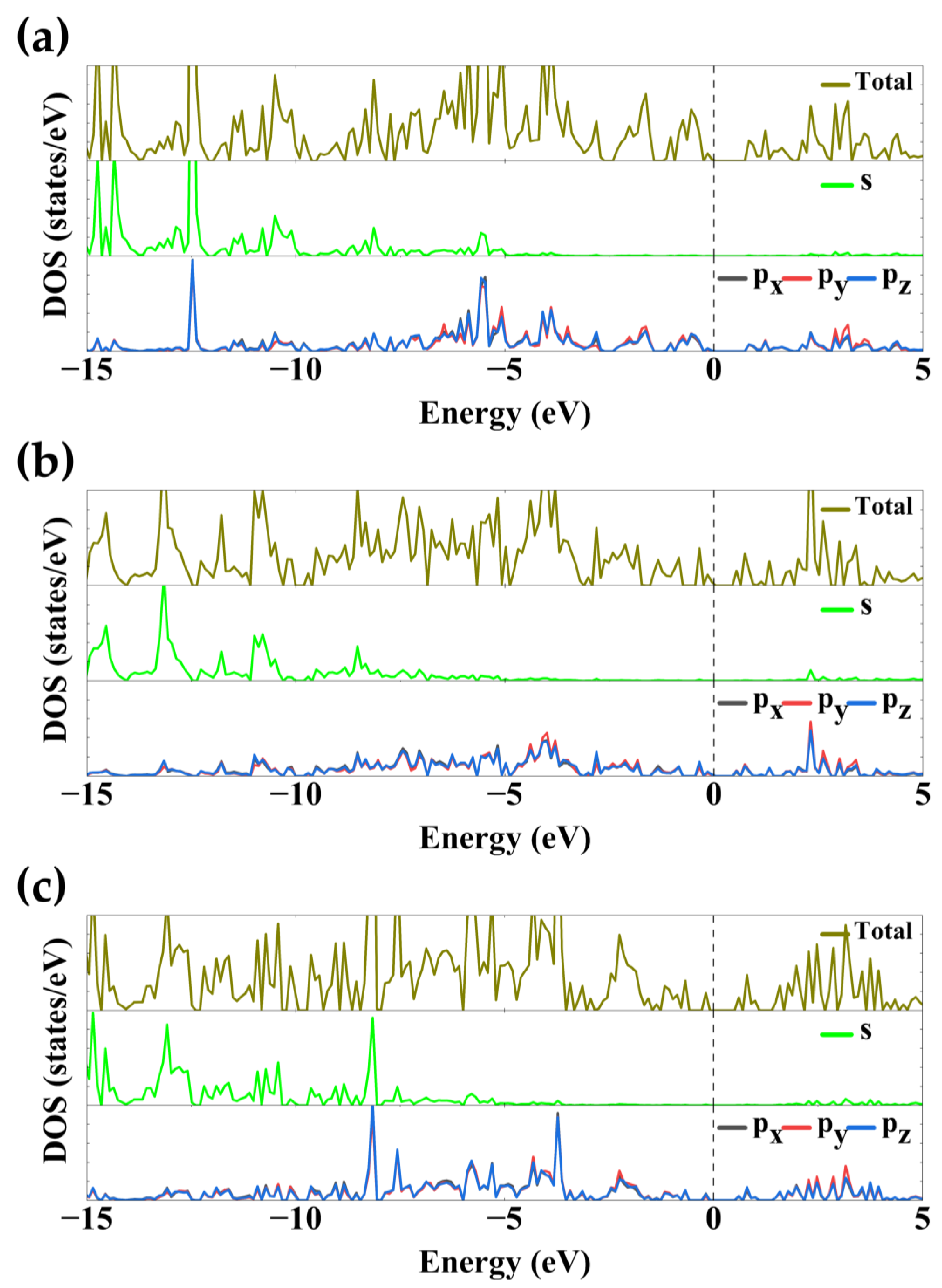
3.6. DOS Analysis
4. Conclusions
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Wu, J.; Pisula, W.; Muellen, K. Graphenes as Potential Material for Electronics. ChemInform 2007, 107, 718–747. [Google Scholar] [CrossRef]
- Geim, A.K. Graphene: Status and Prospects. Science 2009, 324, 1530–1534. [Google Scholar] [CrossRef] [Green Version]
- Ren, S.; Rong, P.; Yu, Q. Preparations, properties and applications of graphene in functional devices: A concise review. Ceram. Int. 2018, 44, 11940–11955. [Google Scholar] [CrossRef]
- Li, X.; Cai, W.; An, J.; Kim, S.; Nah, J.; Yang, D.; Piner, R.; Velamakanni, A.; Jung, I.; Tutuc, E.; et al. Large-Area Synthesis of High-Quality and Uniform Graphene Films on Copper Foils. Science 2009, 324, 1312–1314. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, J.; Wen, Y.; Guo, Y.; Wu, B.; Huang, L.; Xue, Y.; Geng, D.; Wang, D.; Yu, G.; Liu, Y. Oxygen-Aided Synthesis of Polycrystalline Graphene on Silicon Dioxide Substrates. J. Am. Chem. Soc. 2011, 133, 17548–17551. [Google Scholar] [CrossRef]
- Liu, H.; Liu, Y.; Zhu, D. Chemical doping of graphene. J. Mater. Chem. 2011, 21, 3335–3345. [Google Scholar] [CrossRef]
- Li, X.; Zhu, Y.; Cai, W.; Borysiak, M.; Han, B.; Chen, D.; Piner, R.D.; Colombo, L.; Ruoff, R.S. Transfer of large-area graphene films for high-performance transparent conductive electrodes. Nano Lett. 2009, 9, 4359–4363. [Google Scholar] [CrossRef]
- Bae, S.; Kim, H.; Lee, Y.; Xu, X.; Park, J.; Zheng, Y.; Balakrishnan, J.; Lei, T.; Ri Kim, H.; Song, Y.I.; et al. Roll-to-roll production of 30-inch graphene films for transparent electrodes. Nat. Nanotechnol. 2010, 5, 574–578. [Google Scholar] [CrossRef] [Green Version]
- Zhu, Y.; Murali, S.; Cai, W.; Li, X.; Suk, J.W.; Potts, J.R.; Ruoff, R.S. Graphene and Graphene Oxide: Synthesis, Properties, and Applications. Adv. Mater. 2010, 22, 3906–3924. [Google Scholar] [CrossRef]
- Wang, H.; Cui, L.F.; Yang, Y.; Sanchez, C.H.; Robinson, J.T.; Liang, Y.; Cui, Y.; Dai, H. Mn3O4-graphene hybrid as a high-capacity anode material for lithium ion batteries. J. Am. Chem. Soc. 2010, 132, 13978–13980. [Google Scholar] [CrossRef]
- Zhu, Y.; Murali, S.; Stoller, M.D.; Ganesh, K.J.; Cai, W.; Ferreira, P.J.; Pirkle, A.; Wallace, R.M.; Cychosz, K.A.; Thommes, M.; et al. Carbon-based supercapacitors produced by activation of graphene. Science 2011, 332, 1537–1541. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, J.K.; Smith, K.B.; Hayner, C.M.; Kung, H.H. Silicon nanoparticles–graphene paper composites for Li ion battery anodes. Chem. Commun. 2010, 46, 2025–2027. [Google Scholar] [CrossRef]
- Novoselov, K.S.; Geim, A.K.; Morozov, S.V.; Jiang, D.; Zhang, Y.; Dubonos, S.V.; Grigorieva, I.V.; Firsov, A.A. Electric Field Effect in Atomically Thin Carbon Films. Science 2004, 306, 666–669. [Google Scholar] [CrossRef] [Green Version]
- Frindt, R.F. Single Crystals of MoS2 Several Molecular Layers Thick. J. Appl. Phys. 1966, 37, 1928–1929. [Google Scholar] [CrossRef]
- Joensen, P.; Frindt, R.F.; Morrison, S.R. Single-layer MoS2. Mater. Res. Bull. 1986, 21, 457–461. [Google Scholar] [CrossRef]
- Novoselov, K.; Jiang, D.; Schedin, F.; Booth, T.; Khotkevich, V.V.; Morozov, S.; Geim, A. Two Dimentional Atomic Crystals. Proc. Natl. Acad. Sci. USA 2005, 102, 10451. [Google Scholar] [CrossRef] [Green Version]
- Wang, R.; Peng, D.; Hu, J.; Zong, L.; Chen, X. Orientational ordering and electron-phonon interaction in K3C60 superconductor. Carbon 2022, 195, 1–8. [Google Scholar] [CrossRef]
- Zong, L.; Wang, R.; Peng, D.; Chen, X. Superconductivity in Nonstoichiometric Rubidium-Doped C60. J. Phys. Chem. C 2022, 126, 2912–2919. [Google Scholar] [CrossRef]
- Wang, R.; Peng, D.; Zong, L.; Chen, L.; Chen, X. Variation of the critical temperature with the lattice parameter in K3C60. Carbon 2022, 199, 181–188. [Google Scholar] [CrossRef]
- Wang, R.; Peng, D.; Zong, L.; Zhu, Z.; Chen, X. Full set of superconducting parameters of K3C60. Carbon 2023, 202, 325–335. [Google Scholar] [CrossRef]
- Hou, L.; Cui, X.; Guan, B.; Wang, S.; Li, R.; Liu, Y.; Zhu, D.; Zheng, J. Synthesis of a monolayer fullerene network. Nature 2022, 606, 507–510. [Google Scholar] [CrossRef] [PubMed]
- Dungey, K.E.; Curtis, M.D.; Hahn, J.E.P. Structural characterization and thermal stability of MoS2 intercalation compounds. Chem. Mater. 1998, 10, 2152–2161. [Google Scholar] [CrossRef]
- Ouyang, T.; Chen, Y.; Xie, Y.; Yang, K.; Bao, Z.; Zhong, J. Thermal transport in hexagonal boron nitride nanoribbons. Nanotechnology 2010, 21, 245701. [Google Scholar] [CrossRef]
- Tang, S.; Lan, H.; Duan, L.; Jin, J.; Li, J.; Liu, Z.; Wang, G. Co-Precipitation Behavior in Ferrite Region During Isothermal Process in Ti-Mo-Cu Microalloyed Steel. Acta Metall. Sin. 2022, 58, 355–364. [Google Scholar] [CrossRef]
- Radisavljevic, B.; Kis, A. Mobility engineering and a metal-insulator transition in monolayer MoS₂. Nat. Mater. 2013, 12, 815–820. [Google Scholar] [CrossRef]
- Kiriya, D.; Tosun, M.; Zhao, P.; Kang, J.S.; Javey, A. Air-Stable Surface Charge Transfer Doping of MoS2 by Benzyl Viologen. J. Am. Chem. Soc. 2014, 136, 7853–7856. [Google Scholar] [CrossRef]
- Yang, S.; Park, S.; Jang, S.; Kim, H.; Kwon, J. Electrical stability of multilayer MoS2 field-effect transistor under negative bias stress at various temperatures. Phys. Status Solidi Rapid Res. Lett. 2014, 8, 714–718. [Google Scholar] [CrossRef]
- Fan, X.; Zheng, W.T.; Kuo, J.L.; Singh, D.J. Structural stability of single-layer MoS2 under large strain. J. Phys. Condens. Matter 2015, 27, 105401. [Google Scholar] [CrossRef]
- Sen, D.; Das, B.K.; Saha, S.; Roy, R.; Mitra, A.; Chattopadhyay, K.K. sp(3) bonded 2-dimensional allotrope of carbon: A first-principles prediction. Carbon 2019, 146, 430–437. [Google Scholar] [CrossRef]
- Li, L.H.; Cervenka, J.; Watanabe, K.; Taniguchi, T.; Chen, Y. Strong oxidation resistance of atomically thin boron nitride nanosheets. ACS Nano 2014, 8, 1457–1462. [Google Scholar] [CrossRef]
- Wen, B.; Takami, S.; Kawazoe, Y.; Adschiri, T. Pressure-dependent mechanical stability of simple cubic carbon. Phys. B Condens. Matter 2011, 406, 2654–2657. [Google Scholar] [CrossRef]
- Lu, Y.; Zhu, X.; Wang, M. Theoretical investigations of a new two-dimensional carbon allotrope: hP-C23-2D. Comput. Mater. Sci. 2019, 167, 8–12. [Google Scholar] [CrossRef]
- He, C.; Sun, L.; Zhang, C.; Zhong, J. Two viable three-dimensional carbon semiconductors with an entirely sp2 configuration. Phys. Chem. Chem. Phys. 2013, 15, 680–684. [Google Scholar] [CrossRef] [PubMed]
- Luo, Y.; Ren, C.; Xu, Y.; Yu, J.; Wang, S.; Sun, M. A first principles investigation on the structural, mechanical, electronic, and catalytic properties of biphenylene. Sci. Rep. 2021, 11, 19008. [Google Scholar] [CrossRef] [PubMed]
- Mortazavi, B.; Zhuang, X. Low and Anisotropic Tensile Strength and Thermal Conductivity in the Single-Layer Fullerene Network Predicted by Machine-Learning Interatomic Potentials. Coatings 2022, 12, 1171. [Google Scholar] [CrossRef]
- Mei, H.; Zhong, Y.; He, D.; Du, X.; Li, C.; Cheng, N. Predicting the structural, elastic and electronic properties of new two-dimensional carbon and silicon monolayers. Results Phys. 2020, 16, 102826. [Google Scholar] [CrossRef]
- Alborznia, H.; Naseri, M.; Fatahi, N. Pressure effects on the optical and electronic aspects of T-Carbon: A first principles calculation. Optik 2019, 180, 125–133. [Google Scholar] [CrossRef]
- Wang, Y.; Zeng, Q.; Du, X.; Gao, Y.; Yin, B. The structural, mechanical and electronic properties of novel superhard carbon allotropes: Ab initio study. Mater. Today Commun. 2021, 29, 102980. [Google Scholar] [CrossRef]
- Kresse, G.; Furthmüller, J. Efficient Iterative Schemes for Ab Initio Total-Energy Calculations Using a Plane-Wave Basis Set. Phys. Rev. B 1996, 54, 11169. [Google Scholar] [CrossRef]
- Blöchl, P.E. Projector augmented-wave method. Phys. Rev. B 1994, 50, 17953–17979. [Google Scholar] [CrossRef]
- Burke, K.; Ernzerhof, M.; Perdew, J.P. Generalized Gradient Approximation Made Simple. Phys. Rev. Lett. 1996, 77, 3865–3868. [Google Scholar] [CrossRef] [Green Version]
- Tang, S.; Li, L.-X.; Peng, Q.; Yan, H.-L.; Cai, M.-H.; Li, J.-P.; Liu, Z.-Y.; Wang, G.-D. First-principles insights into hydrogen trapping in interstitial-vacancy complexes in vanadium carbide. Phys. Chem. Chem. Phys. 2022, 24, 20400–20408. [Google Scholar] [CrossRef] [PubMed]
- Occelli, F.; Loubeyre, P.; LeToullec, R. Properties of diamond under hydrostatic pressures up to 140 GPa. Nat. Mater. 2003, 2, 151–154. [Google Scholar] [CrossRef] [PubMed]
- Gao, Y.; Feng, X.; Gong, B.; Zhong, C.; Yang, S.A.; Liu, K.; Lu, Z. Theoretical design of all-carbon networks with intrinsic magnetism. Carbon 2021, 177, 11–18. [Google Scholar] [CrossRef]
- Wang, J.; Mizuseki, H.; Chen, C.; Li, Z. Computational discovery of a new rhombohedral diamond phase. Phys. Rev. B 2018, 98, 094107. [Google Scholar] [CrossRef] [Green Version]
- Vejpravová, J. Mixed sp2–sp3 Nanocarbon Materials: A Status Quo Review. Nanomaterials 2021, 11, 2469. [Google Scholar] [CrossRef]
- Popov, A.A.; Yang, S.; Dunsch, L. Endohedral fullerenes. Chem. Rev. 2013, 113, 5989–6113. [Google Scholar] [CrossRef]
- Wang, Q.; Jiang, J. An overview on structure and field emission properties of carbon nitride films. J. Nanomater. 2014, 2014, 10. [Google Scholar] [CrossRef] [Green Version]
- Hu, Q.M.; Yang, R. Mechanical properties of structural materials from first-principles. Curr. Opin. Solid State Mater. Sci. 2006, 10, 19–25. [Google Scholar] [CrossRef]
- Michel, K.H.; Verberck, B. Theory of the elastic constants of graphite and graphene. Phys Status Solidi B 2008, 245, 2177–2180. [Google Scholar] [CrossRef]
- Sun, Y.W.; Papageorgiou, D.G.; Humphreys, C.J.; Dunstan, D.J.; Puech, P.; Proctor, J.E.; Bousige, C.; Machon, D.; San-Miguel, A. Mechanical properties of graphene. Appl. Phys. Rev. 2021, 8, 021310. [Google Scholar] [CrossRef]
- Ramdas, A.K.; Grimsditch, M.H. Brillouin scattering in diamond. Phys. Rev. B 1975, 11, 3139–3148. [Google Scholar] [CrossRef]
- Zhao, S.; Zhang, X.; Ni, Y.; Peng, Q.; Wei, Y. Anisotropic mechanical response of a 2D covalently bound fullerene lattice. Carbon 2022, 202, 118–124. [Google Scholar] [CrossRef]
- Fang, W.; Li, P.; Yuan, J.; Xue, K.; Wang, J. Nb2SiTe4 and Nb2GeTe4: Unexplored 2D Ternary Layered Tellurides with High Stability, Narrow Band Gap and High Electron Mobility. J. Electron. Mater. 2020, 49, 959–968. [Google Scholar] [CrossRef]
- Pugh, S.F. XCII. Relations between the elastic moduli and the plastic properties of polycrystalline pure metals. Lond. Edinb. Dublin Philos. Mag. J. Sci. 1954, 45, 823–843. [Google Scholar] [CrossRef]
- Chen, H.; Cao, Y.; Liu, K.; Tao, X.; Zhou, Y.; Ouyang, Y.; Gao, F.; Du, Y.; Peng, Q. Stability and physical properties tuning via interstitials chemical engineering of Zr5Sn3: A first-principles study. J. Mater. Sci. 2019, 54, 10284–10296. [Google Scholar] [CrossRef]
- Mannepalli, S.; Mangalampalli, K. Indentation Plasticity and Fracture Studies of Organic Crystals. Crystals 2017, 7, 324. [Google Scholar] [CrossRef] [Green Version]
- Chen, X.; Niu, H.; Li, D.; Li, Y. Modeling hardness of polycrystalline materials and bulk metallic glasses. Intermetallics 2011, 19, 1275–1281. [Google Scholar] [CrossRef] [Green Version]
- Tian, Y.; Xu, B.; Zhao, Z. Microscopic theory of hardness and design of novel superhard crystals. Int. J. Refract. Hard Met. 2012, 33, 93–106. [Google Scholar] [CrossRef]
- Jiang, X.; Zhao, J.; Aimin, W.; Bai, Y.; Jiang, X. Mechanical and electronic properties of B-12-based ternary crystals of orthorhombic phase. J. Phys. Condens. Matter 2010, 22, 315503. [Google Scholar] [CrossRef]
- Sayers, C.M.; Kachanov, M. Microcrack-induced elastic wave anisotropy of brittle rocks. J. Geophys. Res. Solid Earth 1995, 100, 4149–4156. [Google Scholar] [CrossRef]
- Gorenstein, M.I.; Zozulya, O.S.; Begun, V.V. Fluctuations in the canonical ensemble. Phys. Rev. C 2005, 72, 014902. [Google Scholar] [CrossRef] [Green Version]
- Singh, N.J.; Yi, H.B.; Min, S.K.; Park, M.; Kim, K.S. Dissolution nature of cesium fluoride by water molecules. J. Phys. Chem. B 2006, 110, 3808–3815. [Google Scholar] [CrossRef] [PubMed]
- Dronskowski, R.; Bloechl, P.E. Crystal orbital Hamilton populations (COHP): Energy-resolved visualization of chemical bonding in solids based on density-functional calculations. J. Phys. Chem. A 1993, 97, 8617–8624. [Google Scholar] [CrossRef]
- Gu, X.J.; Poon, S.J.; Shiflet, G.J.; Widom, M. Ductility improvement of amorphous steels: Roles of shear modulus and electronic structure. Acta Mater. 2008, 56, 88–94. [Google Scholar] [CrossRef]
- Xiao, H.; Tahir-Kheli, J.; Goddard, W.A.I. Accurate Band Gaps for Semiconductors from Density Functional Theory. J. Phys. Chem. Lett. 2011, 2, 212–217. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | a | b | c | Bond Lengths | ΔH | ΔE | Ref. |
|---|---|---|---|---|---|---|---|
| Graphite | 2.47 | 2.27 | 6.74 | 1.42 | −9.20 | Present | |
| 2.46 | 2.46 | 6.70 | 1.42 | [43] | |||
| Graphene | 2.47 | 2.47 | - | 1.42 | 0.05 | −9.15 | Present |
| 2.47 | 2.47 | - | 1.42 | −9.22 | [44] | ||
| Diamond | 3.57 | 3.57 | 3.57 | 1.55 | 0.27 | −8.93 | Present |
| 3.57 | 3.57 | 3.57 | 1.55 | −9.09 | [45] | ||
| qhp-C60 | 9.15 | 9.15 | 7.00 | 1.41–1.64 | 0.45 | −8.75 | Present |
| Compound | C11 | C22 | C33 | C44 | C66 | C12 | B | G | E | v | B/G | Ref. |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| GPa a, N/m b | Dimensionless | |||||||||||
| Graphite | 1093.56 | 33.23 | 4.30 | 428.01 | 212.78 | 161.99 | 115.46 | 279.89 | 0.21 | 1.40 | Present | |
| 1211.30 | 36.79 | 4.18 | 468.00 | 275.50 | [50] | |||||||
| Graphene | 366.90 | 366.80 | 145.95 | 66.60 | 216.72 | 148.04 | 361.75 | 0.20 | 1.46 | Present | ||
| 372.00 | 47.00 | [51] | ||||||||||
| Diamond | 1050.73 | 559.93 | 126.62 | 434.66 | 518.49 | 1112.94 | 0.07 | 0.84 | Present | |||
| 1076.40 | 577.40 | 125.20 | [52] | |||||||||
| qhp-C60 | 140.20 | 192.62 | 63.54 | 23.91 | 93.95 | 66.88 | 188.54(max) | 0.19(max) | 1.41 | Present | ||
| 137.30 | 185.80 | 62.00 | 20.02 | [53] | ||||||||
| Compound | Hchen | Htian | Hjiang | HE | HG |
|---|---|---|---|---|---|
| GPa a, N/m b | |||||
| Graphite | 18.66 | 18.07 | 17.76 | 17.02 | 17.53 |
| Graphene | 20.82 | 20.52 | 22.95 | 21.99 | 23.29 |
| Diamond | 92.24 | 93.97 | 70.62 | 67.67 | 88.82 |
| qhp-C60 | 12.69 | 12.24 | 11.96 | 11.46 | 8.93 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shen, G.; Li, L.; Tang, S.; Jin, J.; Chen, X.-J.; Peng, Q. Stability and Elasticity of Quasi-Hexagonal Fullerene Monolayer from First-Principles Study. Crystals 2023, 13, 224. https://doi.org/10.3390/cryst13020224
Shen G, Li L, Tang S, Jin J, Chen X-J, Peng Q. Stability and Elasticity of Quasi-Hexagonal Fullerene Monolayer from First-Principles Study. Crystals. 2023; 13(2):224. https://doi.org/10.3390/cryst13020224
Chicago/Turabian StyleShen, Guichang, Linxian Li, Shuai Tang, Jianfeng Jin, Xiao-Jia Chen, and Qing Peng. 2023. "Stability and Elasticity of Quasi-Hexagonal Fullerene Monolayer from First-Principles Study" Crystals 13, no. 2: 224. https://doi.org/10.3390/cryst13020224