
Influence of Polymeric Blends on Bioceramics of Hydroxyapatite
 , , and
, , and
Abstract
:1. Introduction
2. Materials and Methods
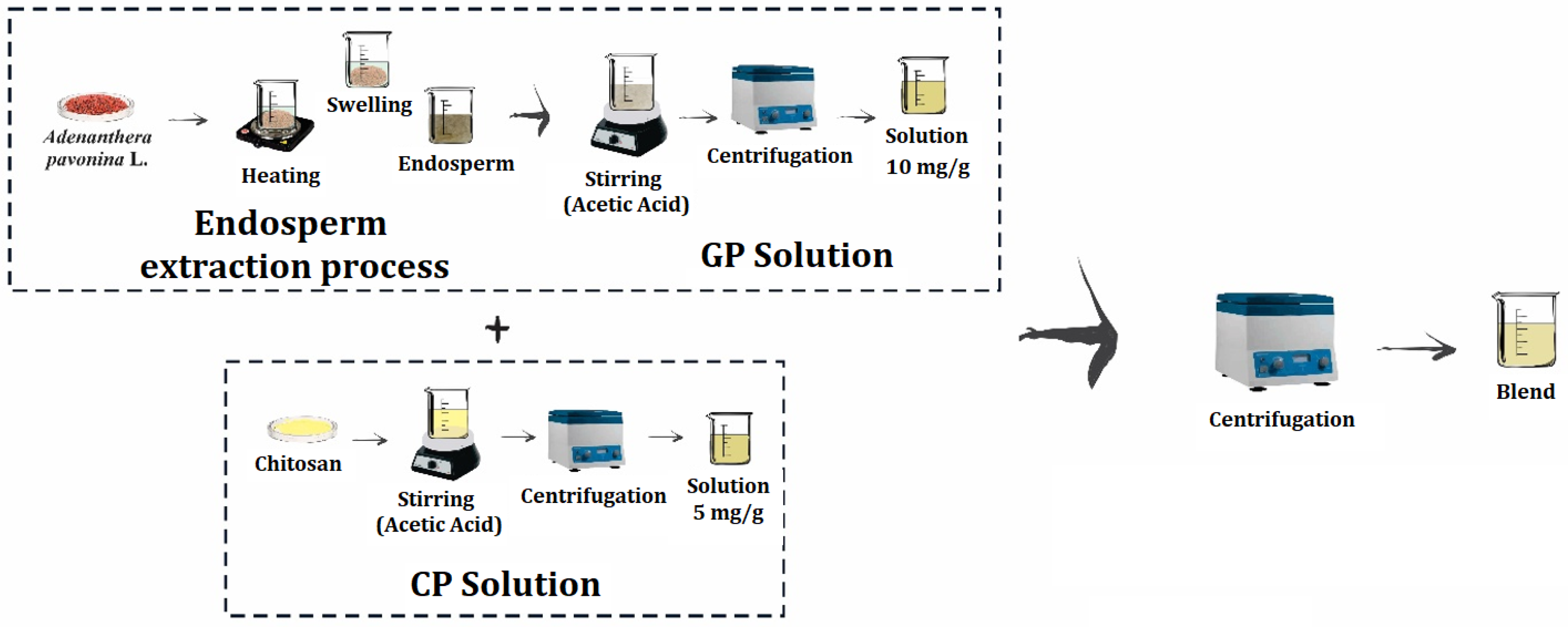
2.1. Bioceramics Preparation
2.2. Bioceramics Characterization
3. Results
3.1. XRD and Rietveld Refinement
3.2. Infrared Spectroscopy
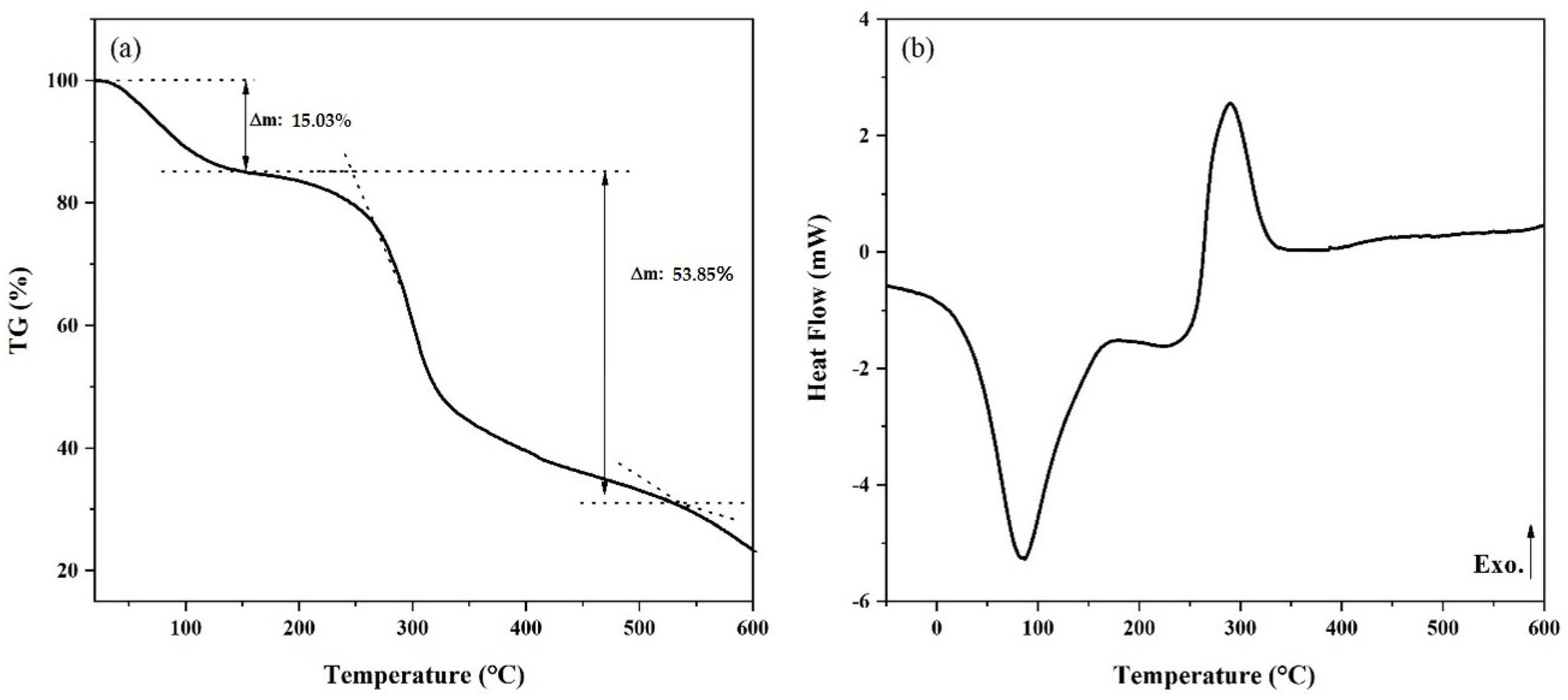
3.3. Thermal Analysis
3.4. Morphological Analysis
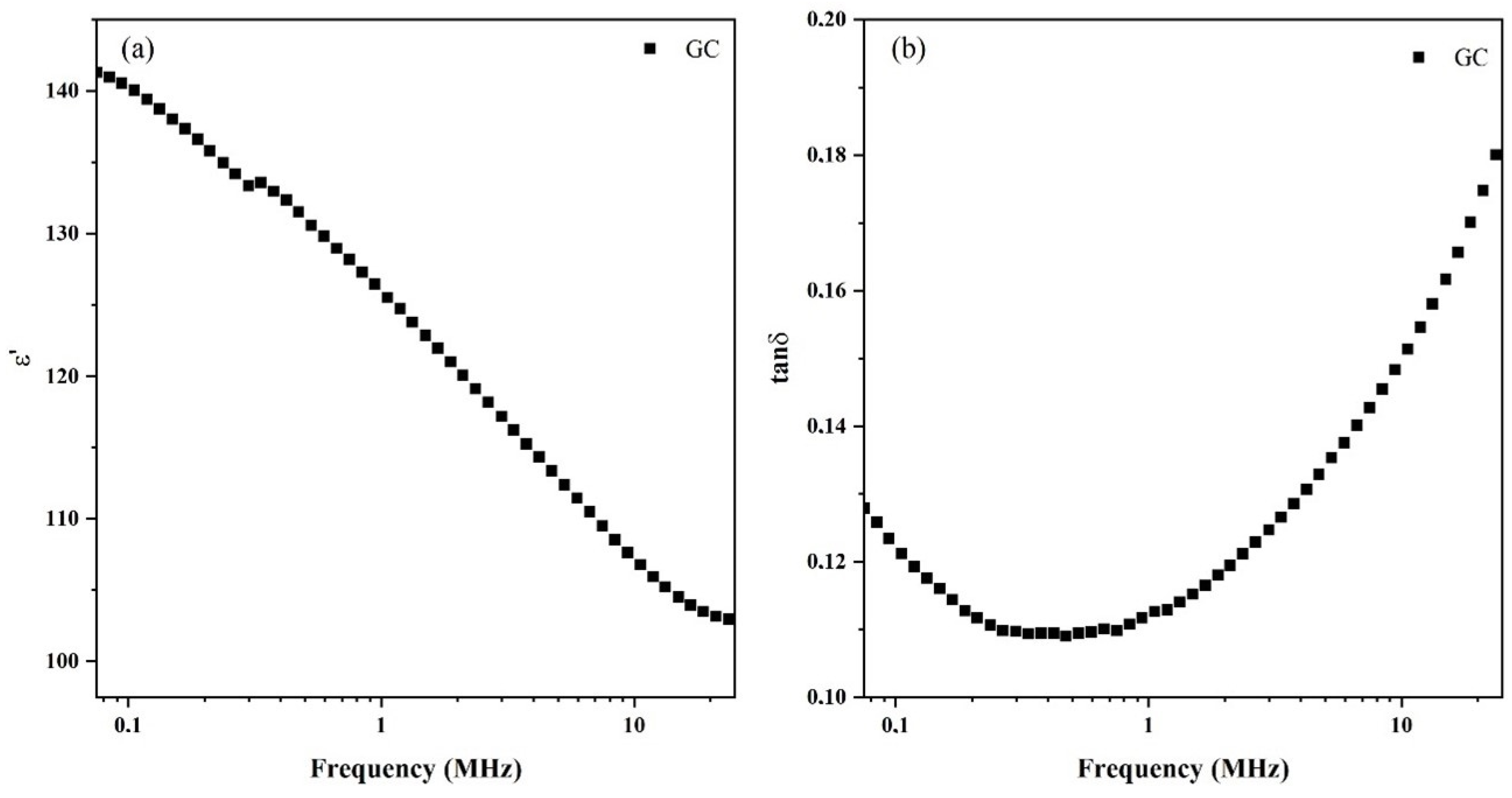
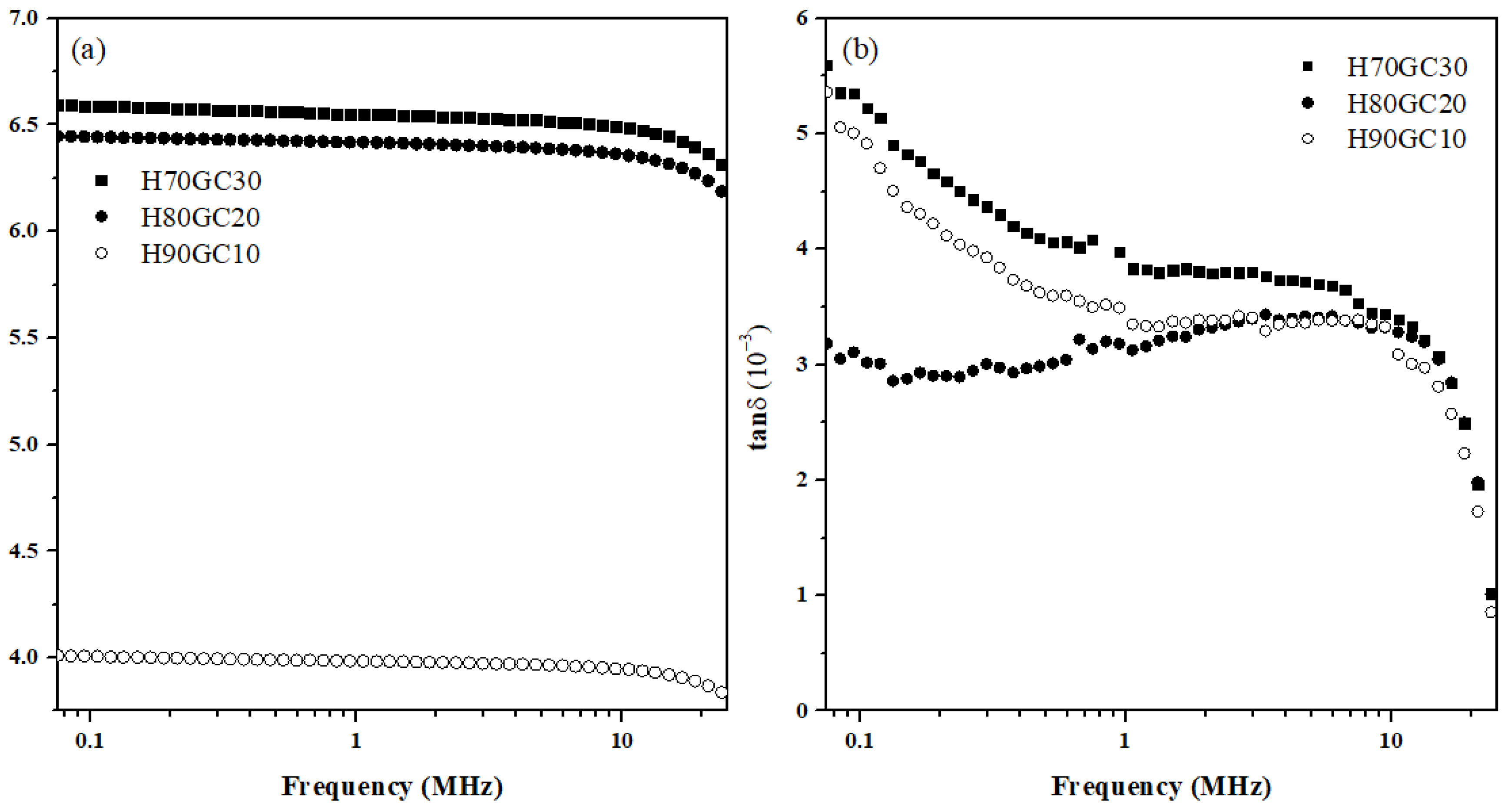
3.5. Dielectric Spectroscopy
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Ragunathan, S.; Govindasamy, G.; Raghul, D.R.; Karuppaswamy, M.; Vijayachandra Togo, R.K. Hydroxyapatite reinforced natural polymer scaffold for bone tissue regeneration. Mater. Today Proc. 2020, 23, 111–118. [Google Scholar] [CrossRef]
- Arvidson, K.; Abdallah, B.M.; Applegate, L.A.; Baldini, N.; Cenni, E.; Gomez-Barrena, E.; Granchi, D.; Kassem, M.; Konttinen, Y.T.; Mustafa, K.; et al. Bone regeneration and stem cells. J. Cell. Mol. Med. 2011, 15, 718–746. [Google Scholar] [CrossRef] [PubMed]
- Kalita, S.J.; Bhardwaj, A.; Bhatt, H.A. Nanocrystalline calcium phosphate ceramics in biomedical engineering. Mater. Sci. Eng. C 2007, 27, 441–449. [Google Scholar] [CrossRef]
- Daglilar, S.; Erkan, M.E. A study on bioceramic reinforced bone cements. Mater. Lett. 2007, 61, 1456–1459. [Google Scholar] [CrossRef]
- Sadat-Shojai, M.; Khorasani, M.T.; Dinpanah-Khoshdargi, E.; Jamshidi, A. Synthesis methods for nanosized hydroxyapatite with diverse structures. Acta Biomater. 2013, 9, 7591–7621. [Google Scholar] [CrossRef]
- Hwang, K.; Lim, Y. Chemical and structural changes of hydroxyapatite films by using a sol–gel method. Surf. Coat. Technol. 1999, 115, 172–175. [Google Scholar] [CrossRef]
- Suchanek, W.L.; Byrappa, K.; Shuk, P.; Riman, R.E.; Janas, V.F.; Ten Huisen, K.S. Preparation of magnesium-substituted hydroxyapatite powders by the mechanochemical–hydrothermal method. Biomaterials 2004, 25, 4647–4657. [Google Scholar] [CrossRef]
- Rigo, E.C.D.S.; Oliveira, L.C.; Santos, L.A.; Boschi, A.O.; Carrodeguas, R.G. Implantes metálicos recobertos com hidroxiapatita. Res. Biomed. Eng. 1999, 15, 21–29. [Google Scholar]
- Assis, C.M.D.; Vercik, L.C.D.O.; Santos, M.L.D.; Fook, M.V.L.; Guastaldi, A.C. Comparison of crystallinity between natural hydroxyapatite and synthetic cp-Ti/HA coatings. Mater. Res. 2005, 8, 207–211. [Google Scholar] [CrossRef]
- Nikpour, M.R.; Rabiee, S.M.; Jahanshahi, M.J.C.P.B.E. Synthesis and characterization of hydroxyapatite/chitosan nanocomposite materials for medical engineering applications. Compos. Part B 2012, 43, 1881–1886. [Google Scholar] [CrossRef]
- Silva, C.C.; Valente, M.A.; Graça, M.P.F.; Sombra, A.S.B. Preparation and optical characterization of hydroxyapatite and ceramic systems with titanium and zirconium formed by dry high-energy mechanical alloying. Solid State Sci. 2004, 6, 1365–1374. [Google Scholar] [CrossRef]
- Motskin, M.; Wright, D.M.; Muller, K.; Kyle, N.; Gard, T.G.; Porter, A.E.; Skepper, J.N. Hydroxyapatite nano and microparticles: Correlation of particle properties with cytotoxicity and biostability. Biomaterials 2009, 30, 3307–3317. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Kempen, D.H.; Yaszemski, M.J.; Lu, L. The roles of matrix polymer crystallinity and hydroxyapatite nanoparticles in modulating material properties of photo-crosslinked composites and bone marrow stromal cell responses. Biomaterials 2009, 30, 3359–3370. [Google Scholar] [CrossRef] [PubMed]
- Mohandes, F.; Salavati-Niasari, M.; Fathi, M.; Fereshteh, Z. Hydroxyapatite nanocrystals: Simple preparation, characterization and formation mechanism. Mater. Sci. Eng. C 2014, 45, 29–36. [Google Scholar] [CrossRef]
- Yasukawa, A.; Kandori, K.; Tanaka, H.; Gotoh, K. Preparation and structure of carbonated calcium hydroxyapatite substituted with heavy rare earth ions. Mater. Res. Bull. 2012, 47, 1257–1263. [Google Scholar] [CrossRef]
- Yasukawa, A.; Gotoh, K.; Tanaka, H.; Kandori, K. Preparation and structure of calcium hydroxyapatite substituted with light rare earth ions. Colloids Surf. A 2012, 393, 53–59. [Google Scholar] [CrossRef]
- Kaygili, O.; Dorozhkin, S.V.; Ates, T.; Al-Ghamdi, A.A.; Yakuphanoglu, F. Dielectric properties of Fe doped hydroxyapatite prepared by sol–gel method. Ceram. Int. 2014, 40, 9395–9402. [Google Scholar] [CrossRef]
- Zhang, W.; Cao, N.; Chai, Y.; Xu, X.; Wang, Y. Synthesis of nanosize single-crystal strontium hydroxyapatite via a simple sol–gel method. Ceram. Int. 2014, 40, 16061–16064. [Google Scholar] [CrossRef]
- Silva, C.C.; Graça, M.P.F.; Valente, M.A.; Sombra, A.S.B. Structural study of Fe2O3-doped calcium phosphates obtained by the mechanical milling method. Phys. Scr. 2009, 79, 055601. [Google Scholar] [CrossRef]
- Nasiri-Tabrizi, B.; Fahami, A.; Ebrahimi-Kahrizsangi, R. Effect of milling parameters on the formation of nanocrystalline hydroxyapatite using different raw materials. Ceram. Inter. 2013, 39, 5751–5763. [Google Scholar] [CrossRef]
- Rhee, S.H. Synthesis of hydroxyapatite via mechanochemical treatment. Biomaterials 2002, 23, 1147–1152. [Google Scholar] [CrossRef] [PubMed]
- Cho, J.S.; Rhee, S.H. Formation mechanism of nano-sized hydroxyapatite powders through spray pyrolysis of a calcium phosphate solution containing polyethylene glycol. J. Eur. Ceram. Soc. 2013, 33, 233–241. [Google Scholar] [CrossRef]
- Liu, H.S.; Chin, T.S.; Lai, L.S.; Chiu, S.Y.; Chung, K.H.; Chang, C.S.; Lui, M.T. Hydroxyapatite synthesized by a simplified hydrothermal method. Ceram. Inter. 1997, 23, 19–25. [Google Scholar] [CrossRef]
- Lala, S.; Satpati, B.; Kar, T.; Pradhan, S.K. Structural and microstructural characterizations of nanocrystalline hydroxyapatite synthesized by mechanical alloying. Mater. Sci. Eng. C 2013, 33, 2891–2898. [Google Scholar] [CrossRef] [PubMed]
- Macuvele, D.L.P.; Nones, J.; Matsinhe, J.V.; Lima, M.M.; Soares, C.; Fiori, M.A.; Riella, H.G. Advances in ultra high molecular weight polyethylene/hydroxyapatite composites for biomedical applications: A brief review. Mater. Sci. Eng. C 2017, 76, 1248–1262. [Google Scholar] [CrossRef] [PubMed]
- Chen, F.; Wang, Z.C.; Lin, C.J. Preparation and characterization of nano-sized hydroxyapatite particles and hydroxyapatite/chitosan nano-composite for use in biomedical materials. Mater. Lett. 2002, 57, 858–861. [Google Scholar] [CrossRef]
- Zhang, L.J.; Feng, X.S.; Liu, H.G.; Qian, D.J.; Zhang, L.; Yu, X.L.; Cui, F.Z. Hydroxyapatite/collagen composite materials formation in simulated body fluid environment. Mater. Lett. 2004, 58, 719–722. [Google Scholar] [CrossRef]
- Maeda, Y.; Jayakumar, R.; Nagahama, H.; Furuike, T.; Tamura, H. Synthesis, characterization and bioactivity studies of novel β-chitin scaffolds for tissue-engineering applications. Int. J. Biol. Macromol. 2008, 42, 463–467. [Google Scholar] [CrossRef]
- Manara, S.; Paolucci, F.; Palazzo, B.; Marcaccio, M.; Foresti, E.; Tosi, G.; Sabbatini, S.; Sabatino, P.; Altankov, G.; Roveri, N. Electrochemically-assisted deposition of biomimetic hydroxyapatite–collagen coatings on titanium plate. Inorg. Chim. Acta 2008, 361, 1634–1645. [Google Scholar] [CrossRef]
- Madhumathi, K.; Shalumon, K.T.; Rani, V.D.; Tamura, H.; Furuike, T.; Selvamurugan, N.; Nair, S.V.; Jayakumar, R. Wet chemical synthesis of chitosan hydrogel–hydroxyapatite composite membranes for tissue engineering applications. Int. J. Biol. Macromol. 2009, 45, 12–15. [Google Scholar] [CrossRef]
- Pang, X.; Casagrande, T.; Zhitomirsky, I. Electrophoretic deposition of hydroxyapatite–CaSiO3–chitosan composite coatings. J. Colloid Interface Sci. 2009, 330, 323–329. [Google Scholar] [CrossRef] [PubMed]
- Trakoolwannachai, V.; Kheolamai, P.; Ummartyotin, S. Development of hydroxyapatite from eggshell waste and a chitosan-based composite: In vitro behavior of human osteoblast-like cell (Saos-2) cultures. Int. J. Biol. Macromol. 2019, 134, 557–564. [Google Scholar] [CrossRef] [PubMed]
- Pereira, M.B.B.; França, D.B.; Araújo, R.C.; Silva Filho, E.C.; Rigaud, B.; Fonseca, M.G.; Jaber, M. Amino hydroxyapatite/chitosan hybrids reticulated with glutaraldehyde at different pH values and their use for diclofenac removal. Carbohydr. Polym. 2020, 236, 116036. [Google Scholar] [CrossRef] [PubMed]
- Macêdo, A.A.M.; Sombra, A.S.B.; Mazzetto, S.E.; Silva, C.C. Influence of the polysaccharide galactomannan on the dielectrical characterization of hydroxyapatite ceramic. Compos. Part B 2013, 44, 95–99. [Google Scholar] [CrossRef]
- Pawlik, A.; Rehman, M.A.U.; Nawaz, Q.; Bastan, F.E.; Sulka, G.D.; Boccaccini, A.R. Fabrication and characterization of electrophoretically deposited chitosan-hydroxyapatite composite coatings on anodic titanium dioxide layers. Electrochim. Acta 2019, 307, 465–473. [Google Scholar] [CrossRef]
- Cunha, C.S.; Castro, P.J.; Sousa, S.C.; Pullar, R.C.; Tobaldi, D.M.; Piccirillo, C.; Pintado, M.M. Films of chitosan and natural modified hydroxyapatite as effective UV-protecting, biocompatible and antibacterial wound dressings. Int. J. Biol. Macromol. 2020, 159, 1177–1185. [Google Scholar] [CrossRef]
- Olivera, S.; Muralidhara, H.B.; Venkatesh, K.; Guna, V.K.; Gopalakrishna, K.; Kumar, Y. Potential applications of cellulose and chitosan nanoparticles/composites in wastewater treatment: A review. Carbohydr. Polym. 2016, 153, 600–618. [Google Scholar] [CrossRef]
- Croisier, F.; Jérôme, C. Chitosan-based biomaterials for tissue engineering. Eur. Polym. J. 2013, 49, 780–792. [Google Scholar] [CrossRef]
- Thanyacharoen, T.; Chuysinuan, P.; Techasakul, S.; Noenplab, A.N.L.; Ummartyotin, S. The chemical composition and antioxidant and release properties of a black rice (Oryza sativa L.)-loaded chitosan and polyvinyl alcohol composite. J. Mol. Liq. 2017, 248, 1065–1070. [Google Scholar] [CrossRef]
- Thanyacharoen, T.; Chuysinuan, P.; Techasakul, S.; Nooeaid, P.; Ummartyotin, S. Development of a gallic acid-loaded chitosan and polyvinyl alcohol hydrogel composite: Release characteristics and antioxidant activity. Int. J. Biol. Macromol. 2018, 107, 363–370. [Google Scholar] [CrossRef]
- Kök, M.S.; Hill, S.E.; Mitchell, J.R. Viscosity of galactomannans during high temperature processing: Influence of degradation and solubilisation. Food Hydrocoll. 1999, 13, 535–542. [Google Scholar] [CrossRef]
- Fernandes, P.B.; Gonçalves, M.P.; Doublier, J.L. A rheological characterization of kappa-carrageenan/galactomannan mixed gels: A comparison of locust bean gum samples. Carbohydr. Polym. 1991, 16, 253–274. [Google Scholar] [CrossRef]
- Bresolin, T.M.B.; Milas, M.; Rinaudo, M.; Reicher, F.; Ganter, J.L.M.S. Role of galactomannan composition on the binary gel formation with xanthan. Int. J. Biol. Macromol. 1999, 26, 225–231. [Google Scholar] [CrossRef] [PubMed]
- Vendruscolo, C.W.; Andreazza, I.F.; Ganter, J.L.M.S.; Ferrero, C.; Bresolin, T.M.B. Xanthan and galactomannan (from M. scabrella) matrix tablets for oral controlled delivery of theophylline. Int. J. Pharm. 2005, 296, 1–11. [Google Scholar] [CrossRef]
- Rietveld, H.M. Line profiles of neutron powder-diffraction peaks for structure refinement. Acta Crystallogr. 1967, 22, 151–152. [Google Scholar] [CrossRef]
- Doebelin, N.; Kleeberg, R. Profex: A graphical user interface for the Rietveld refinement program BGMN. J. Appl. Crystallogr. 2015, 48, 1573–1580. [Google Scholar] [CrossRef]
- Hayakawa, S.; Li, Y.; Tsuru, K.; Osaka, A.; Fujii, E.; Kawabata, K. Preparation of nanometer-scale rod array of hydroxyapatite crystal. Acta Biomater. 2009, 5, 2152–2160. [Google Scholar] [CrossRef]
- Vinodhini, P.A.; Sangeetha, K.; Thandapani, G.; Sudha, P.N.; Jayachandran, V.; Sukumaran, A. FTIR, XRD and DSC studies of nanochitosan, cellulose acetate and polyethylene glycol blend ultrafiltration membranes. Int. J. Biol. Macromol. 2017, 104, 1721–1729. [Google Scholar] [CrossRef]
- Hoepfner, T.P.; Case, E.D. The influence of the microstructure on the hardness of sintered hydroxyapatite. Ceram. Int. 2003, 29, 699–706. [Google Scholar] [CrossRef]
- Pinto Filho, F.; Nogueira, R.E.F.Q.; Graça, M.P.F.; Valente, M.A.; Sombra, A.S.B.; Silva, C.C.D. Structural and mechanical study of the sintering effect in hydroxyapatite doped with iron oxide. Physica B 2008, 403, 3826–3829. [Google Scholar] [CrossRef]
- Figueiró, S.D.; Macêdo, A.A.M.; Melo, M.R.S.; Freitas, A.L.P.; Moreira, R.A.; De Oliveira, R.S.; Goés, J.C.; Sombra, A.S.B. On the dielectric behaviour of collagen–algal sulfated polysaccharide blends: Effect of glutaraldehyde crosslinking. Biophys. Chem. 2006, 120, 154–159. [Google Scholar] [CrossRef] [PubMed]
- Peixoto, M.V.; Costa, F.M.; Devesa, S.; Graça, M.P.F. Structural, Morphological and Dielectric Characterization of BiFeO3 Fibers Grown by the LFZ Technique. Crystals 2023, 13, 960. [Google Scholar] [CrossRef]
- Ashoorirad, M.; Saviz, M.; Fallah, A. On the electrical properties of collagen macromolecule solutions: Role of collagen-water interactions. J. Mol. Liq. 2020, 300, 112344. [Google Scholar] [CrossRef]
- Salema, A.A.; Yeow, Y.K.; Ishaque, K.; Ani, F.N.; Afzal, M.T.; Hassan, A. Dielectric properties and microwave heating of oil palm biomass and biochar. Ind. Crops Prod. 2013, 50, 366–374. [Google Scholar] [CrossRef]
- Dorey, R. Microstructure–property relationships: How the microstructure of the film affects its properties. In Ceramic Thick Films for MEMS and Microdevices; Dorey, R., Ed.; William Andrew Publishing: Norwich, NY, USA, 2012; pp. 85–112. [Google Scholar] [CrossRef]
- Graça, M.P.F.; da Silva, M.F.; Valente, M.A. NaNbO3 crystals dispersed in a B2O3 glass matrix–Structural characteristics versus electrical and dielectrical properties. Solid State Sci. 2009, 11, 570–577. [Google Scholar] [CrossRef]
- Kazin, P.E.; Pogosova, M.A.; Trusov, L.A.; Kolesnik, I.V.; Magdysyuk, O.V.; Dinnebier, R.E. Crystal structure details of La-and Bi-substituted hydroxyapatites: Evidence for LaO+ and BiO+ with a very short metal–oxygen bond. J. Solid State Chem. 2016, 237, 349–357. [Google Scholar] [CrossRef]
- Silva, C.C.; Pinheiro, A.G.; Miranda, M.A.R.; Góes, J.C.; Sombra, A.S.B. Structural properties of hydroxyapatite obtained by mechanosynthesis. Solid State Sci. 2013, 5, 553–558. [Google Scholar] [CrossRef]
- Sarkar, A.; Kannan, S. In situ synthesis, fabrication and Rietveld refinement of the hydroxyapatite/titania composite coatings on 316 L SS. Ceram. Int. 2014, 40, 6453–6463. [Google Scholar] [CrossRef]
- Bhadang, K.A.; Gross, K.A. Influence of fluorapatite on the properties of thermally sprayed hydroxyapatite coatings. Biomaterials 2004, 25, 4935–4945. [Google Scholar] [CrossRef]
- Silva, C.C.D.; Rocha, H.H.B.; Freire, F.N.A.; Santos, M.R.P.; Sabóia, K.D.A.; Góes, J.C.; Sombra, A.S.B. Hydroxyapatite screen-printed thick films: Optical and electrical properties. Mater. Chem. Phys. 2005, 92, 260–268. [Google Scholar] [CrossRef]
- Sanosh, K.P.; Chu, M.C.; Balakrishnan, A.; Lee, Y.J.; Kim, T.N.; Cho, S.J. Synthesis of nano hydroxyapatite powder that simulate teeth particle morphology and composition. Curr. Appl. Phys. 2009, 9, 1459–1462. [Google Scholar] [CrossRef]
- Xia, Z.; Liao, L.; Zhao, S. Synthesis of mesoporous hydroxyapatite using a modified hard-templating route. Mater. Res. Bull. 2009, 44, 1626–1629. [Google Scholar] [CrossRef]
- Yang, P.; Quan, Z.; Li, C.; Kang, X.; Lian, H.; Lin, J. Bioactive, luminescent and mesoporous europium-doped hydroxyapatite as a drug carrier. Biomaterials 2008, 29, 4341–4347. [Google Scholar] [CrossRef]
- Wang, J.; Somasundaran, P. Study of galactomannose interaction with solids using AFM, IR and allied techniques. J. Colloid Interface Sci. 2007, 309, 373–383. [Google Scholar] [CrossRef] [PubMed]
- Tang, E.S.K.; Huang, M.; Lim, L.Y. Ultrasonication of chitosan and chitosan nanoparticles. Int. J. Pharm 2003, 265, 103–114. [Google Scholar] [CrossRef]
- Ding, W.; Lian, Q.; Samuels, R.J.; Polk, M.B. Synthesis and characterization of a novel derivative of chitosan. Polymer 2003, 44, 547–556. [Google Scholar] [CrossRef]
- Govindan, S.; Nivethaa, E.A.K.; Saravanan, R.; Narayanan, V.; Stephen, A. Synthesis and characterization of chitosan–silver nanocomposite. Appl. Nanosci. 2012, 2, 299–303. [Google Scholar] [CrossRef]
- Lima, C.G.A.; De Oliveira, R.S.; Figueiró, S.D.; Wehmann, C.F.; Góes, J.C.; Sombra, A.S.B. DC conductivity and dielectric permittivity of collagen–chitosan films. Mater. Chem. Phys. 2016, 99, 284–288. [Google Scholar] [CrossRef]
- Figueiró, S.D.; Góes, J.C.; Moreira, R.A.; Sombra, A.S.B. On the physico-chemical and dielectric properties of glutaraldehyde crosslinked galactomannan–collagen films. Carbohydr. Polym. 2004, 56, 313–320. [Google Scholar] [CrossRef]
- Park, S.H.; Chun, M.K.; Choi, H.K. Preparation of an extended-release matrix tablet using chitosan/Carbopol interpolymer complex. Int. J. Pharm. 2018, 347, 39–44. [Google Scholar] [CrossRef]
- Mandal, T.; Mishra, B.K.; Garg, A.; Chaira, D. Optimization of milling parameters for the mechanosynthesis of nanocrystalline hydroxyapatite. Powder Technol. 2014, 253, 650–656. [Google Scholar] [CrossRef]
- Bulina, N.V.; Makarova, S.V.; Baev, S.G.; Matvienko, A.A.; Gerasimov, K.B.; Logutenko, O.A.; Bystrov, V.S. A study of thermal stability of hydroxyapatite. Minerals 2021, 11, 1310. [Google Scholar] [CrossRef]
- Tonsuaadu, K.; Gross, K.A.; Plūduma, L.; Veiderma, M. A review on the thermal stability of calcium apatites. J. Therm. Anal. Calorim. 2012, 110, 647–659. [Google Scholar] [CrossRef]
- Wang, T.; Dorner-Reisel, A.; Müller, E. Thermogravimetric and thermokinetic investigation of the dehydroxylation of a hydroxyapatite powder. J. Eur. Ceram. Soc. 2004, 24, 693–698. [Google Scholar] [CrossRef]
- Mostafa, N.Y. Characterization, thermal stability and sintering of hydroxyapatite powders prepared by different routes. Mater. Chem. Phys. 2005, 94, 333–341. [Google Scholar] [CrossRef]
- Zima, A. Hydroxyapatite-chitosan based bioactive hybrid biomaterials with improved mechanical strength. Spectrochim. Acta Part A 2018, 193, 175–184. [Google Scholar] [CrossRef]
- Rahman, P.M.; Mujeeb, V.A.; Muraleedharan, K.; Thomas, S.K. Chitosan/nano ZnO composite films: Enhanced mechanical, antimicrobial and dielectric properties. Arabian J. Chem. 2018, 11, 120–127. [Google Scholar] [CrossRef]
- Raja, V.; Sharma, A.K.; Rao, V.N. Impedance spectroscopic and dielectric analysis of PMMA-CO-P4VPNO polymer films. Mater. Lett. 2004, 58, 3242–3247. [Google Scholar] [CrossRef]
- Bonardd, S.; Robles, E.; Barandiaran, I.; Saldías, C.; Leiva, Á.; Kortaberria, G. Biocomposites with increased dielectric constant based on chitosan and nitrile-modified cellulose nanocrystals. Carbohydr. Polym. 2018, 199, 20–30. [Google Scholar] [CrossRef]
- Al-Muntaser, A.A.; Pashameah, R.A.; Alzahrani, E.; AlSubhi, S.A.; Hameed, S.T.; Morsi, M.A. Graphene nanoplatelets/TiO2 hybrid nanofiller boosted PVA/CMC blend based high performance nanocomposites for flexible energy storage applications. J. Polym. Environ. 2023, 31, 2534–2548. [Google Scholar] [CrossRef]
- El-Naggar, A.M.; Heiba, Z.K.; Kamal, A.M.; Alzahrani, K.E.; Abd-Elkader, O.H.; Mohamed, M.B. Impact of natural melanin doping on the structural, optical and dielectric characteristics of the PVP/CMC blend. J. Taibah Univ. Sci. 2023, 17, 2190731. [Google Scholar] [CrossRef]
- Abdullah, A.Q.; Ali, N.A.; Hussein, S.I.; Hakamy, A.; Abd-Elnaiem, A.M. Improving the Dielectric, Thermal, and Electrical Properties of Poly (Methyl Methacrylate)/Hydroxyapatite Blends by Incorporating Graphene Nanoplatelets. J. Inorg. Organomet. Polym. Mater. 2023, 1–12. [Google Scholar] [CrossRef]
- Bhatt, A.S.; Bhat, D.K.; Santosh, M.S. Electrical and magnetic properties of chitosan-magnetite nanocomposites. Physica B 2010, 405, 2078–2082. [Google Scholar] [CrossRef]
- Costa, M.M.; Sohn, R.S.T.M.; Macêdo, A.A.M.; Mazzetto, S.E.; Graça, M.P.F.; Sombra, A.S.B. Study of the temperature and organic bindings effects in the dielectric and structural properties of the lithium ferrite ceramic matrix (LiFe5O8). J. Alloys Compd. 2011, 509, 9466–9471. [Google Scholar] [CrossRef]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | Lattice Parameter | RWP (%) | S | ρ (g/cm3) | LC (nm) | XC (%) | ||
|---|---|---|---|---|---|---|---|---|
| a = b (Å) | c (Å) | V (Å3) | ||||||
| HAP | 9.4204 (7) | 6.8823 (5) | 528.9358 (5) | 15.01 | 1.08 | 3.147 | 30.45 ± 0.7 | 37.52 |
| Bands | Wavenumber (cm−1) | |
|---|---|---|
| HAP | GC | |
| υ2 (O-P-O) | 478 | - |
| υ4 (O-P-O) | 569 | - |
| υ4 (O-P-O) | 603 | - |
| OH− | 630 | - |
| α–D–galactopyranose | - | 808 |
| β–D–mannopyranose | - | 879 |
| υ1 (P-O) | 968 | - |
| CH3 | - | 1139 |
| υ3 (P-O) | 1043 | - |
| υ3 (P-O) | 1093 | - |
| C-O-C stretching vibration | 1000 to 1110 | |
| Symmetric deformations of groups CH2 and COH | - | 1350 to 1450 |
| υ3 (CO32−) | 1419 | - |
| υ3 (CO32−) | 1469 | - |
| N−H | - | 1575 |
| OH− | 1649 | - |
| C=O | - | 1660 |
| Bioceramics | Average Grain Size (μm) | Density (g/cm3) | Vickers Hardness (GPa) |
|---|---|---|---|
| HAP | 0.5 | 2.61 | 1.9 |
| H70GC30 | 0.75 | 2.20 | 0.73 |
| H80GC20 | 0.69 | 2.32 | 0.79 |
| H90GC10 | 0.60 | 2.67 | 0.97 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gomes, E.d.S.; Lima, A.M.d.O.; Gavinho, S.R.; Graça, M.P.F.; Devesa, S.; Macêdo, A.A.M. Influence of Polymeric Blends on Bioceramics of Hydroxyapatite. Crystals 2023, 13, 1429. https://doi.org/10.3390/cryst13101429
Gomes EdS, Lima AMdO, Gavinho SR, Graça MPF, Devesa S, Macêdo AAM. Influence of Polymeric Blends on Bioceramics of Hydroxyapatite. Crystals. 2023; 13(10):1429. https://doi.org/10.3390/cryst13101429
Chicago/Turabian StyleGomes, Eduardo da Silva, Antônia Millena de Oliveira Lima, Sílvia Rodrigues Gavinho, Manuel Pedro Fernandes Graça, Susana Devesa, and Ana Angélica Mathias Macêdo. 2023. "Influence of Polymeric Blends on Bioceramics of Hydroxyapatite" Crystals 13, no. 10: 1429. https://doi.org/10.3390/cryst13101429