
Probing Optoelectronic and Thermoelectric Properties of Lead-Free Perovskite SnTiO3: HSE06 and Boltzmann Transport Calculations

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Computational Details
3. Results and Discussion
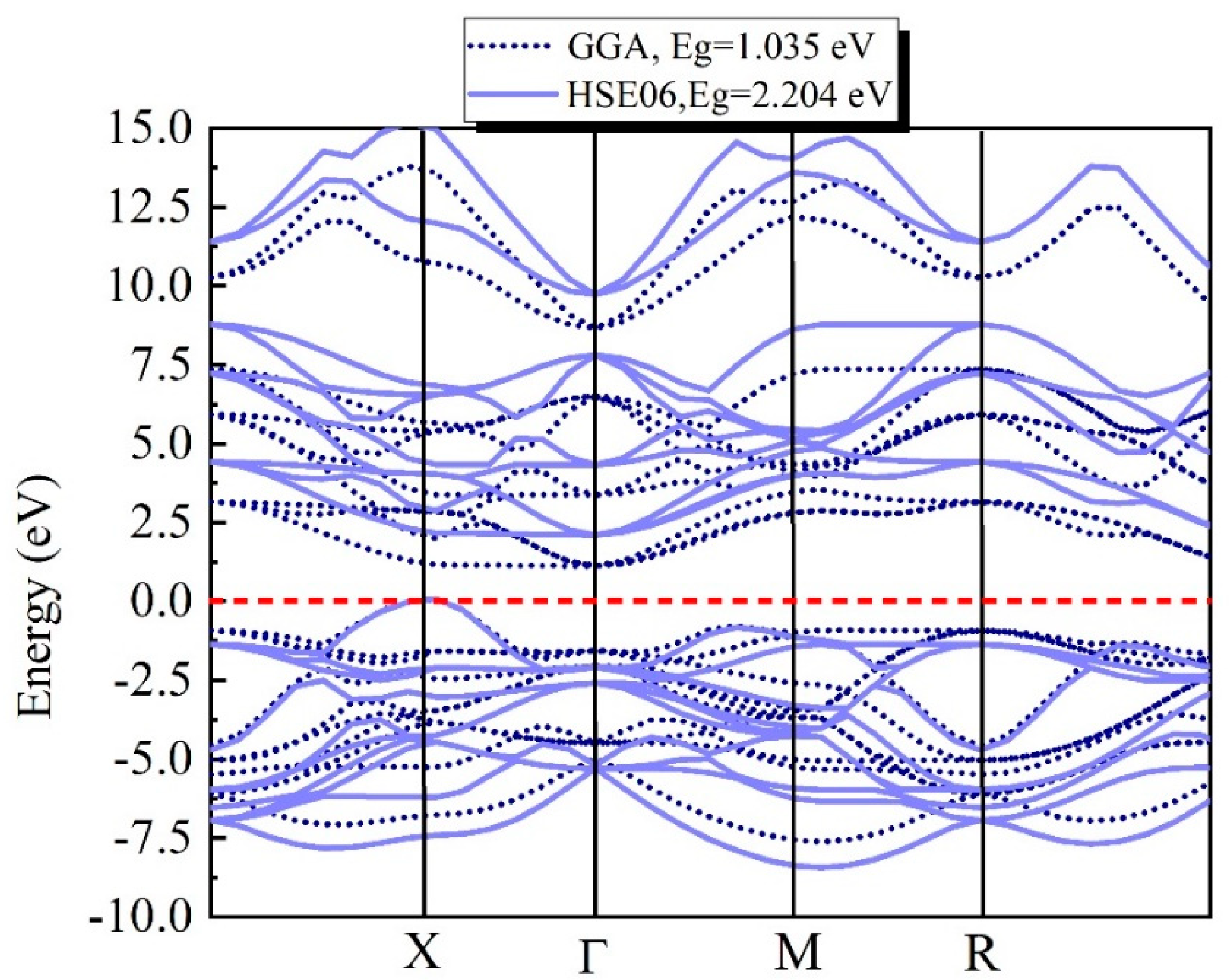
3.1. Electronic Structure Calculations
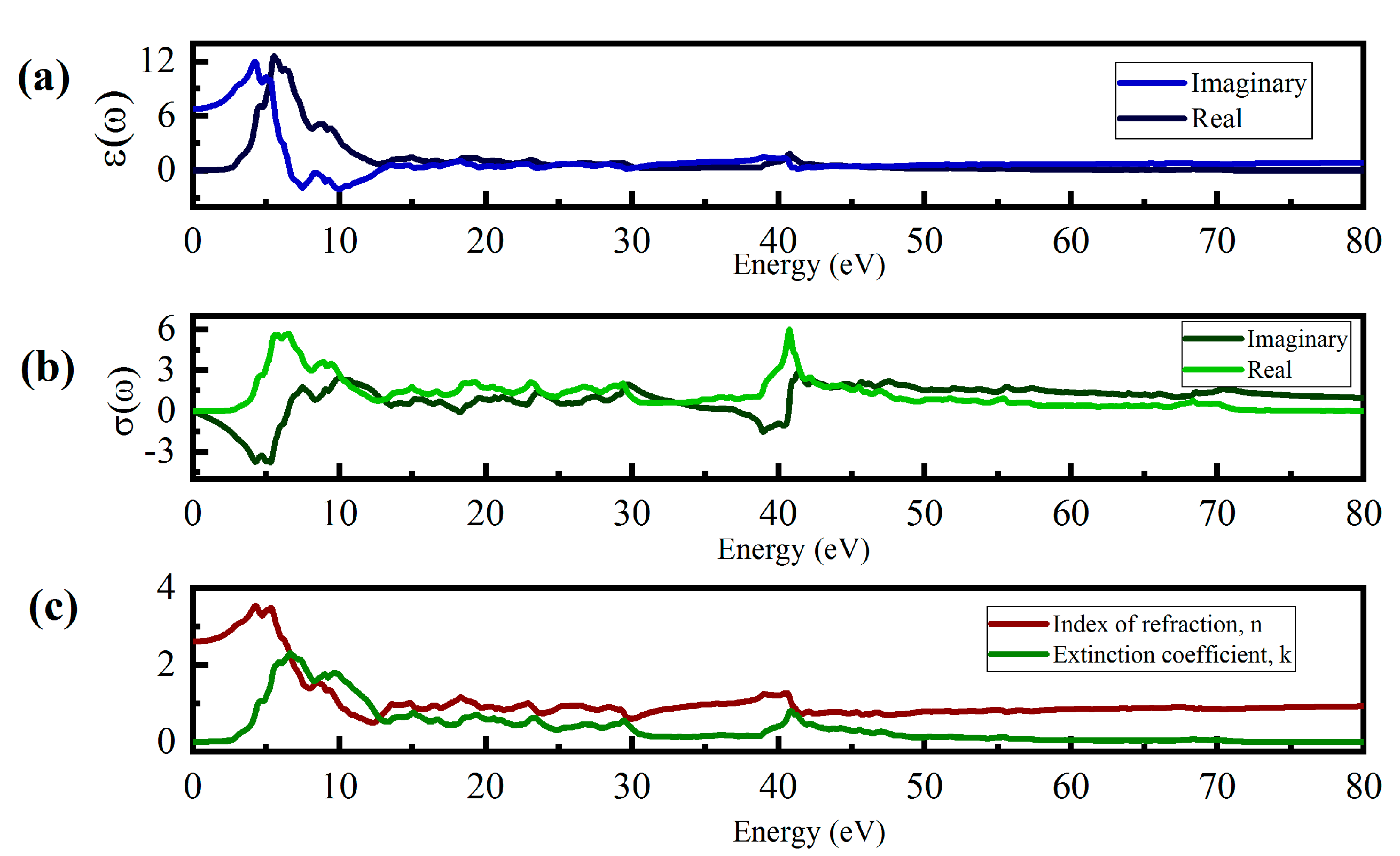
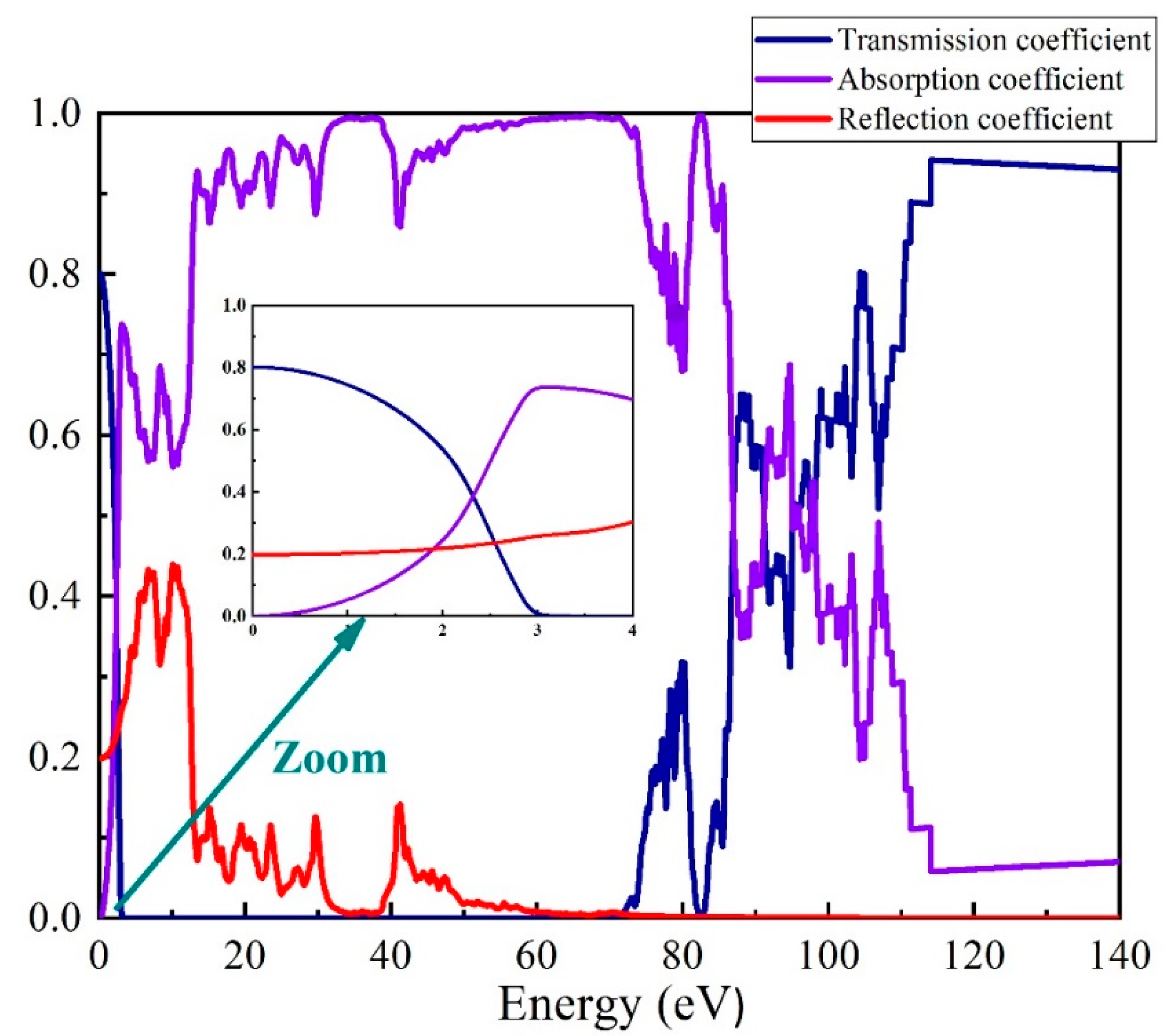
3.2. Optical Properties
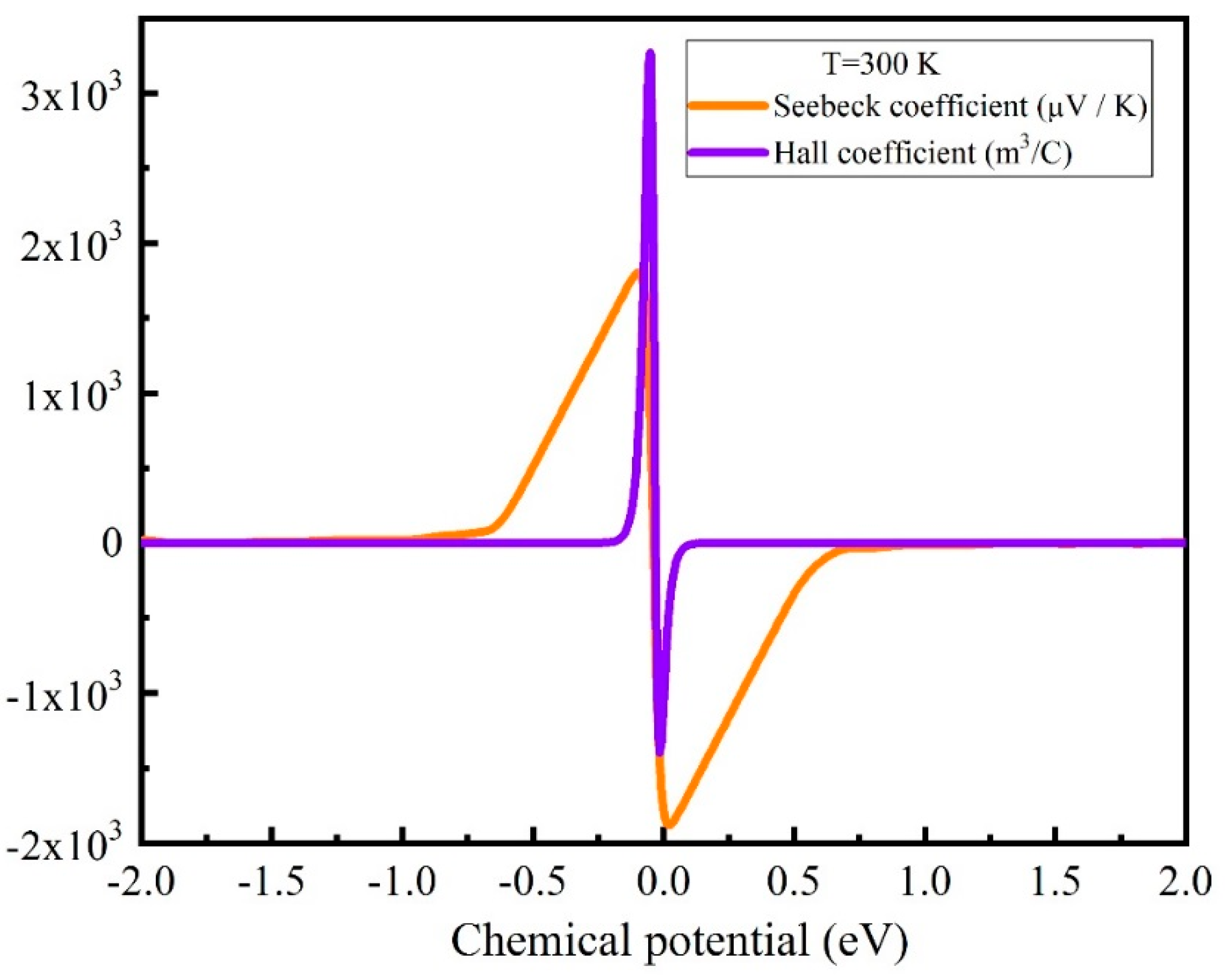
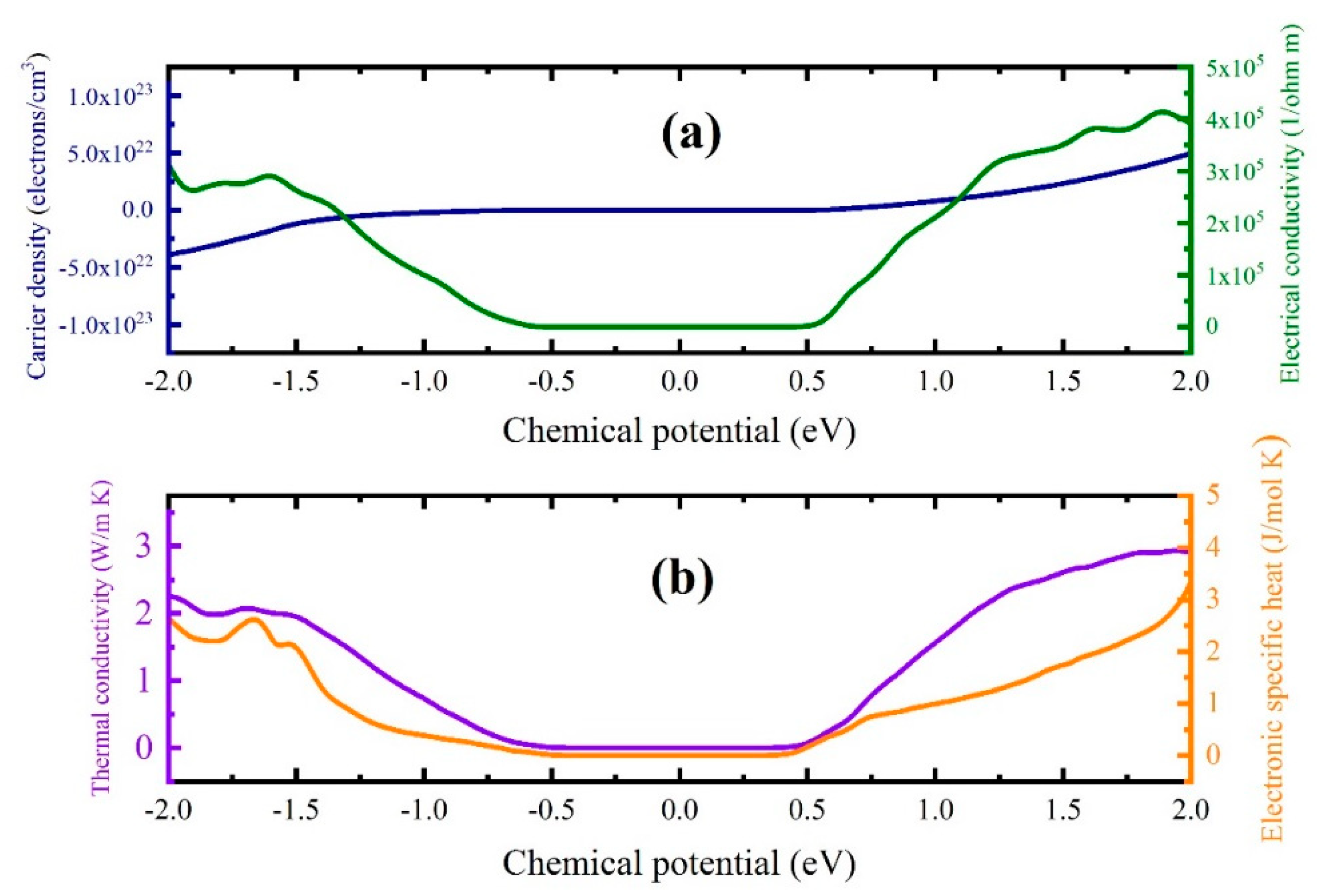
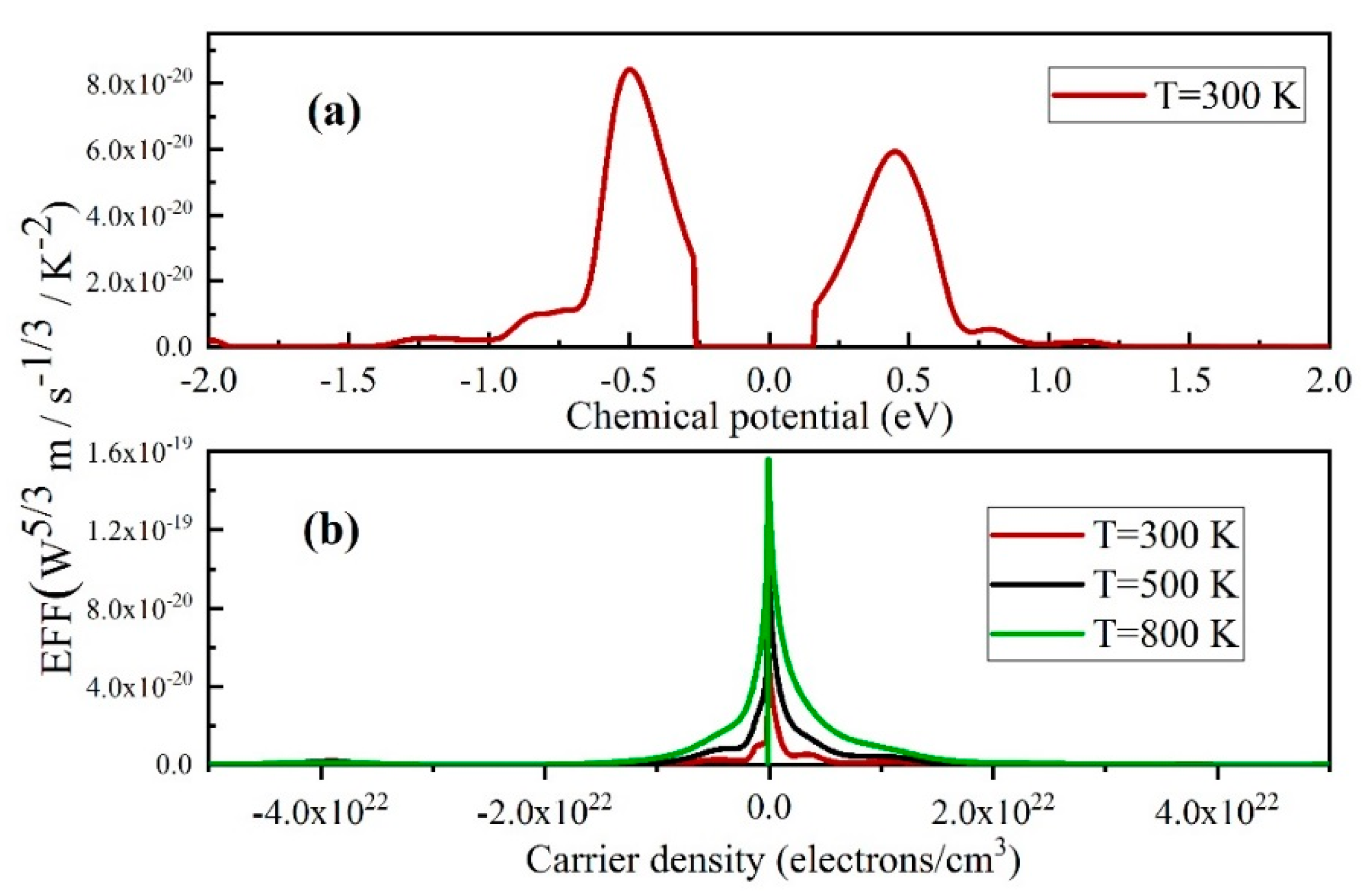
3.3. Boltzmann Transport Calculations and Thermoelectric Properties
4. Conclusions
Funding
Data Availability Statement
Conflicts of Interest
References
- Im, J.-H.; Lee, C.-R.; Lee, J.-W.; Park, S.-W.; Park, N.-G. 6.5% efficient perovskite quantum-dot-sensitized solar cell. Nanoscale 2011, 3, 4088–4093. [Google Scholar] [CrossRef] [PubMed]
- Moon, S.-J.; Itzhaik, Y.; Yum, J.-H.; Zakeeruddin, S.M.; Hodes, G.; Grätzel, M.J. Sb2S3-Based Mesoscopic Solar Cell using an Organic Hole Conductor. Phys. Chem. Lett. 2010, 1, 1524–1527. [Google Scholar] [CrossRef]
- Kanoun, M.B.; Goumri-Said, S. Insights into the impact of Mn-doped inorganic CsPbBr3 perovskite on electronic structures and magnetism for photovoltaic application. Mater. Today Energy 2021, 21, 100796, ISSN 2468-6069. [Google Scholar] [CrossRef]
- Ali, N.; Shehzad, N.; Uddin, S.; Ahmed, R.; Jabeen, M.; Kalam, A.; Al-Sehemi, A.G.; Alrobei, H.; Kanoun, M.B.; Khesro, A.; et al. A review on perovskite materials with solar cell prospective. Int. J. Energy Res. 2021, 45, 19729–19745. [Google Scholar] [CrossRef]
- Fadla, M.A.; Bentria, B.; Benghia, A.; Dahame, T.; Goumri-Said, S. Insights on the opto-electronic structure of the inorganic mixed halide perovskites γ-CsPb(I1−xBrx)3 with low symmetry black phase. J. Alloys Compd. 2020, 832, 154847, ISSN 0925-8388. [Google Scholar] [CrossRef]
- Akkerman, Q.A.; Gandini, M.; di Stasio, F.; Rastogi, P.; Palazon, F.; Bertoni, G.; Ball, J.M.; Prato, M.; Petrozza, A.; Manna, L. Strongly emissive perovskite nanocrystal inks for high-voltage solar cells. Nat. Energy 2016, 2, 16194. [Google Scholar] [CrossRef]
- Eperon, G.E.; Paternò, G.M.; Sutton, R.J.; Zampetti, A.; Haghighirad, A.A.; Cacialli, F.; Snaith, H.J. Inorganic caesium lead iodide perovskite solar cells. J. Mater. Chem. A 2015, 3, 19688–19695. [Google Scholar] [CrossRef]
- Zhang, P.; Ochi, T.; Fujitsuka, M.; Kobori, Y.; Majima, T.; Tachikawa, T. Topotactic Epitaxy of SrTiO3 Mesocrystal Superstructures with Anisotropic Construction for Efficient Overall Water Splitting. Angew. Chem. Int. Ed. 2017, 56, 5299–5303. [Google Scholar] [CrossRef]
- Dynys, F.W.; Berger, M.-H.; Sehirlioglu, A. Thermoelectric Properties of Undoped and Doped (Ti0.75Sn0.25)O2. J. Am. Ceram. Soc. 2012, 95, 619–626. [Google Scholar] [CrossRef]
- Moore, L.A.; Smith, C.M. Reduced Strontium Titanate Thermoelectric Materials. In Ceramics for Environmental and Energy Applications II; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2014; pp. 43–55. [Google Scholar]
- Xie, F.; Zhang, L.; Su, D.; Jaroniec, M.; Qiao, S.-Z. Na2Ti3O7@N-Doped Carbon Hollow Spheres for Sodium-Ion Batteries with Excellent Rate Performance. Adv. Mater. 2017, 29, 1700989. [Google Scholar] [CrossRef]
- Dos santos-Garcia, A.J.; Solana-Madruga, E.; Ritter, C.; Avila-Brande, D.; Fabelo, O.; Saez-Puche, R. Synthesis, structures and magnetic properties of the dimorphic Mn2CrSbO6 oxide. Dalton Trans. 2015, 44, 10665–10672. [Google Scholar] [CrossRef] [PubMed]
- Wahila, M.J.; Butler, K.T.; Lebens-Higgins, Z.W.; Hendon, C.H.; Nandur, A.S.; Treharne, R.E.; Quackenbush, N.F.; Sallis, S.; Mason, K.; Paik, H.; et al. Lone-Pair Stabilization in Transparent Amorphous Tin Oxides: A Potential Route to p-Type Conduction Pathways. Chem. Mater. 2016, 28, 4706–4713. [Google Scholar] [CrossRef]
- Walsh, A.; Payne, D.J.; Egdell, R.G.; Watson, G.W. Stereochemistry of post-transition metal oxides: Revision of the classical lone pair model. Chem. Soc. Rev. 2011, 40, 4455–4463. [Google Scholar] [CrossRef]
- Lotsch, B.V. Ein Klassiker im neuen Gewand: Perowskit-Solarzellen. Angew. Chem. 2014, 126, 647–649. [Google Scholar] [CrossRef]
- Lermer, C.; Birkhold, S.T.; Moudrakovski, I.L.; Mayer, P.; Schoop, L.M.; Schmidt-Mende, L.; Lotsch, B.V. Toward Fluorinated Spacers for MAPI-Derived Hybrid Perovskites: Synthesis, Characterization, and Phase Transitions of (FC2H4NH3)2PbCl4. Chem. Mater. 2016, 28, 6560–6566. [Google Scholar] [CrossRef]
- de Lazaro, S.; Longo, E.; Sambrano, J.R.; Beltrán, A. Structural and electronic properties of PbTiO3 slabs: A DFT periodic study. Surf. Sci. 2004, 552, 149–159. [Google Scholar] [CrossRef]
- Konishi, Y.; Ohsawa, M.; Yonezawa, Y.; Tanimura, Y.; Chikyow, T.; Wakisaka, T.; Koinuma, H.; Miyamoto, A.; Kubo, M.; Sasata, K. Possible Ferroelectricity in SnTiO3 by First-Principles Calculations. MRS Online Proceedings Library (OPL). 2002, Volume 748. Available online: https://www.cambridge.org/core/journals/mrs-online-proceedings-library-archive/article/abs/possible-ferroelectricity-in-sntio3-by-firstprinciples-calculations/DFE24DBF7BF2418A257BBB98519599A7 (accessed on 10 June 2022).
- Piskunov, S.; Heifets, E.; Eglitis, R.I.; Borstel, G. Bulk properties and electronic structure of SrTiO3, BaTiO3, PbTiO3 perovskites: An ab initio HF/DFT study. Comput. Mater. Sci. 2004, 29, 165–178. [Google Scholar] [CrossRef]
- Jain, A.; Ong, S.; Hautier, G.; Chen, W.; Richards, W.; Dacek, S.; Cholia, S.; Gunter, D.; Skinner, D.; Ceder, G.; et al. Commentary: The Materials Project: A materials genome approach to accelerating materials innovation. APL Mater. 2013, 1, 011002. [Google Scholar] [CrossRef]
- Woods-Robinson, R.; Han, Y.; Zhang, H.; Ablekim, T.; Khan, I.; Persson, K.; Zakutayev, A. Wide Band Gap Chalcogenide Semiconductors. Chem. Rev. 2020, 120, 4007–4055. [Google Scholar] [CrossRef]
- Zhou, J.; Sumpter, B.; Kent, P.; Huang, J. A Novel and Functional Single-Layer Sheet of ZnSe. ACS Appl. Mater. Interfaces 2015, 7, 1458–1464. [Google Scholar] [CrossRef]
- Li, W.; Walther, C.; Kuc, A.; Heine, T. Density Functional Theory and Beyond for Band-Gap Screening: Performance for Transition-Metal Oxides and Dichalcogenides. J. Chem. Theory Comput. 2013, 9, 2950–2958. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Xi, L.; Wang, Y.; Zhang, J.; Zhang, P.; Zhang, W. Electronic properties of energy harvesting Cu-chalcogenides: P–d hybridization and d-electron localization. Comput. Mater. Sci. 2015, 108, 239–249. [Google Scholar] [CrossRef]
- Brandbyge, M.; Mozos, J.L.; Ordejon, P.; Taylor, J.; Stokbro, K. Density-functional method for nonequilibrium electron transport. Phys. Rev. B 2002, 65, 165401. [Google Scholar] [CrossRef]
- Soler, J.M.; Artacho, E.; Gale, J.D.; García, A.; Junquera, J.; Ordejón, P.; Sánchez-Portal, D. The SIESTA method for ab initio order-N materials simulation. J. Phys. Condens. Matter 2002, 14, 2745. [Google Scholar] [CrossRef]
- Becke, A.D. A new mixing of hartree–fock and local density-functional theories. J. Chem. Phys. 1993, 98, 1372–1377. [Google Scholar] [CrossRef]
- Heyd, J.; Scuseria, G.E.; Ernzerhof, M. Hybrid functionals based on a screened coulomb potential. J. Chem. Phys. 2003, 118, 8207–8215. [Google Scholar] [CrossRef]
- Heyd, J.; Scuseria, G.E.; Ernzerhof, M. Erratum: “Hybrid functionals based on a screened Coulomb potential” [J. Chem. Phys. 118, 8207 (2003). J. Chem. Phys. 2006, 124, 219906. [Google Scholar] [CrossRef]
- Liu, D.C.; Nocedal, J. On the limited memory BFGS method for large scale optimization. Math. Program. 1989, 45, 503–528. [Google Scholar] [CrossRef]
- Raghupathy, R.K.M.; Wiebeler, H.; Kuhne, T.D.; Felser, C.; Mirhosseini, H. Database Screening of Ternary Chalcogenides for P-type Transparent Conductors. Chem. Mater. 2018, 30, 6794–6800. [Google Scholar] [CrossRef]
- Gonze, X.; Lee, C. Dynamical matrices, born effective charges, dielectric permittivity tensors, and interatomic force constants from density-functional perturbation theory. Phys. Rev. B 1997, 55, 10355. [Google Scholar] [CrossRef]
- Scheidemantel, T.J.; Ambrosch-Draxl, C.; Thonhauser, T.; Badding, J.V.; Sofo, J.O. Transport coefficients from first-principles calculations. Phys. Rev. B 2003, 68, 125210. [Google Scholar] [CrossRef]
- Park, M.S.; Song, J.-H.; Medvedeva, J.E.; Kim, M.; Kim, I.G.; Freeman, A.J. Electronic structure and volume effect on thermoelectric transport in p-type Bi and Sb tellurides. Phys. Rev. B 2010, 81, 155211. [Google Scholar] [CrossRef]
- Madsen, G.K.H.; Singh, D.J. BoltzTraP. A code for calculating band-structure dependent quantities. Comput. Phys. Commun. 2006, 175, 67–71. [Google Scholar] [CrossRef]
- Yang, J.; Li, H.; Wu, T.; Zhang, W.; Chen, L.; Yang, J. Evaluation of Half-Heusler Compounds as Thermoelectric Materials Based on the Calculated Electrical Transport Properties. Adv. Funct. Mater. 2008, 18, 2880–2888. [Google Scholar] [CrossRef]
- Fei, R.; Faghaninia, A.; Soklaski, R.; Yan, J.-A.; Lo, C.; Yang, L. Enhanced Thermoelectric Efficiency via Orthogonal Electrical and Thermal Conductances in Phosphorene. Nano Lett. 2014, 14, 6393–6399. [Google Scholar] [CrossRef] [PubMed]
- Bjerg, L.; Madsen, G.K.H.; Iversen, B.B. Ab initio Calculations of Intrinsic Point Defects in ZnSb. Chem. Mater. 2011, 23, 3907. [Google Scholar] [CrossRef]
- Sun, J.; Singh, D.J. Thermoelectric properties of n-type SrTiO3. APL Mater. 2016, 4, 104803. [Google Scholar] [CrossRef]
- Madsen, G.K.H. Automated Search for New Thermoelectric Materials: The Case of LiZnSb. J. Am. Chem. Soc. 2006, 128, 12140–12146. [Google Scholar] [CrossRef]
- Pulikkotil, J.J.; Singh, D.J.; Auluck, S.; Saravanan, M.; Misra, D.K.; Dhar, A.; Budhani, R.C. Doping and temperature dependence of thermoelectric properties in Mg2(Si,Sn). Phys. Rev. B 2012, 86, 155204. [Google Scholar] [CrossRef]
- Bhattacharya, S.; Madsen, G.K.H. A novel p-type half-Heusler from high-throughput transport and defect calculations. J. Mater. Chem. C 2016, 4, 11261–11268. [Google Scholar] [CrossRef]
- Chen, W.; Pöhls, J.-H.; Hautier, G.; Broberg, D.; Bajaj, S.; Aydemir, U.; Gibbs, Z.M.; Zhu, H.; Asta, M.; Snyder, G.J.; et al. Understanding thermoelectric properties from high-throughput calculations: Trends, insights, and comparisons with experiment. J. Mater. Chem. C 2016, 4, 4414–4426. [Google Scholar] [CrossRef]
- Xing, G.; Sun, J.; Ong, K.P.; Fan, X.; Zheng, W.; Singh, D.J. Perspective: N-type oxide thermoelectrics via visual search strategies. APL Mater. 2016, 4, 053201. [Google Scholar] [CrossRef]
- Sun, J.; Singh, D.J. Thermoelectric properties of AMg2X2, AZn2Sb2 (A = Ca, Sr, Ba; X = Sb, Bi), and Ba2ZnX2 (X = Sb, Bi) Zintl compounds. J. Mater. Chem. A 2017, 5, 8499–8509. [Google Scholar] [CrossRef]
- Taib, M.F.M.; Yaakob, M.K.; Hassan, O.H.; Yahya, M.Z.A. Structural, Electronic, and Lattice Dynamics of PbTiO3, SnTiO3, and SnZrO3: A Comparative First-Principles Study. Integr. Ferroelectr. 2013, 142, 119–127. [Google Scholar] [CrossRef]
- Cardon, F.; Gomes, W.P. On the determination of the flat-band potential of a semiconductor in contact with a metal or an electrolyte from the Mott-Schottky plot. J. Phys. D Appl. Phys. 1978, 11, L63–L67. [Google Scholar] [CrossRef]
- Kalyanasundaram, K.; Grätzel, M. Applications of functionalized transition metal complexes in photonic and optoelectronic devices. Coord. Chem. Rev. 1998, 177, 347–414. [Google Scholar] [CrossRef]
- Alam, N.N.; Malik, N.A.; Hussin, N.H.; Ali, A.M.M.; Hassan, O.H.; Yahya, M.Z.A.; Taib, M.F.M. First-Principles Study on Electronic Properties, Phase Stability and Strain Properties of Cubic (Pm3m) and Tetragonal (P4mm) ATiO3 (A = Pb, Sn). Int. J. Nanoelectron. Mater. 2020, 13, 281–288. [Google Scholar]
- Kittel, C. Introduction to Solid State Physics; Wiley: New York, NY, USA, 1996; ISBN 978-0-471-14286-7. [Google Scholar]
- Li, X.; Xia, C.; Wang, M.; Wu, Y.; Chen, D. First-Principles Investigation of Structural, Electronic and Elastic Properties of HfX (X = Os, Ir and Pt) Compounds. Metals 2017, 7, 317. [Google Scholar] [CrossRef]
- Karki, B.B.; Wentzcovitch, R.M.; de Gironcoli, S.; Baroni, S. High-pressure lattice dynamics and thermoelasticity of MgO. Phys. Rev. B 2000, 61, 8793. [Google Scholar] [CrossRef]
- Amin, B.; Ahmad, I.; Maqbool, M.; Goumri-Said, S.; Ahmad, R. Ab initio study of the bandgap engineering of Al1−xGaxN for optoelectronic applications. J. Appl. Phys. 2011, 109, 023109. [Google Scholar] [CrossRef]
- Singh, D.J.; Mazin, I. Calculated thermoelectric properties of La-filled skutterudites. Phys. Rev. B 1997, 56, R1650. [Google Scholar] [CrossRef] [Green Version]
- Hurd, C.M. The Hall Effect in Metals and Alloys; Plenum Press: New York, NY, USA, 1972. [Google Scholar]
- Onoue, M.; Ishii, F.; Oguchi, T. Electronic and Thermoelectric Properties of the Intermetallic Compounds MNiSn (M = Ti, Zr, and Hf). J. Phys. Soc. Jpn. 2008, 77, 054706. [Google Scholar] [CrossRef]
- Xing, G.; Sun, J.; Li, Y.; Fan, X.; Zheng, W.; Singh, D.J. Electronic fitness function for screening semiconductors as thermoelectric materials. Phys. Rev. Mater. 2017, 1, 65405. [Google Scholar] [CrossRef]
- Feng, Z.; Fu, Y.; Putatunda, A.; Hang, Y.; Singh, D.J. Electronic structure as a guide in screening for potential thermoelectrics: Demonstration for half-Heusler compounds. Phys. Rev. B 2019, 100, 085202. [Google Scholar] [CrossRef]
- Azam, S.; Goumri-Said, S.; Khan, S.A.; Kanoun, M.B. Electronic, optical and thermoelectric properties of new metal-rich homological selenides with palladium–indium: Density functional theory and Boltzmann transport model. J. Phys. Chem. Solids 2020, 138, 109229. [Google Scholar] [CrossRef]
- Azam, S.; Khan, S.A.; Goumri-Said, S. Optoelectronic and Thermoelectric Properties of Bi2OX2 (X = S, Se, Te) for Solar Cells and Thermoelectric Devices. J. Electron. Mater. 2018, 47, 2513–2518. [Google Scholar] [CrossRef]
- Goumri-Said, S.; Alrebdi, T.A.; Deligoz, E.; Ozisik, H.; Kanoun, M.B. Revisiting the Electronic Structures and Phonon Properties of Thermoelectric Antimonide-Tellurides: Spin–Orbit Coupling Induced Gap Opening in ZrSbTe and HfSbTe. Crystals 2021, 11, 917. [Google Scholar] [CrossRef]








Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Goumri-Said, S. Probing Optoelectronic and Thermoelectric Properties of Lead-Free Perovskite SnTiO3: HSE06 and Boltzmann Transport Calculations. Crystals 2022, 12, 1317. https://doi.org/10.3390/cryst12091317
Goumri-Said S. Probing Optoelectronic and Thermoelectric Properties of Lead-Free Perovskite SnTiO3: HSE06 and Boltzmann Transport Calculations. Crystals. 2022; 12(9):1317. https://doi.org/10.3390/cryst12091317
Chicago/Turabian StyleGoumri-Said, Souraya. 2022. "Probing Optoelectronic and Thermoelectric Properties of Lead-Free Perovskite SnTiO3: HSE06 and Boltzmann Transport Calculations" Crystals 12, no. 9: 1317. https://doi.org/10.3390/cryst12091317