

Theoretical Study on the Electrochemical Water Splitting of Two-Dimensional Metal–Organic Frameworks TM3C12O12 (TM = Mn, Fe, Co, Ni)
Abstract
:1. Introduction
2. Computational Details
3. Results
3.1. Geometry and Stability
3.2. Electronic Properties
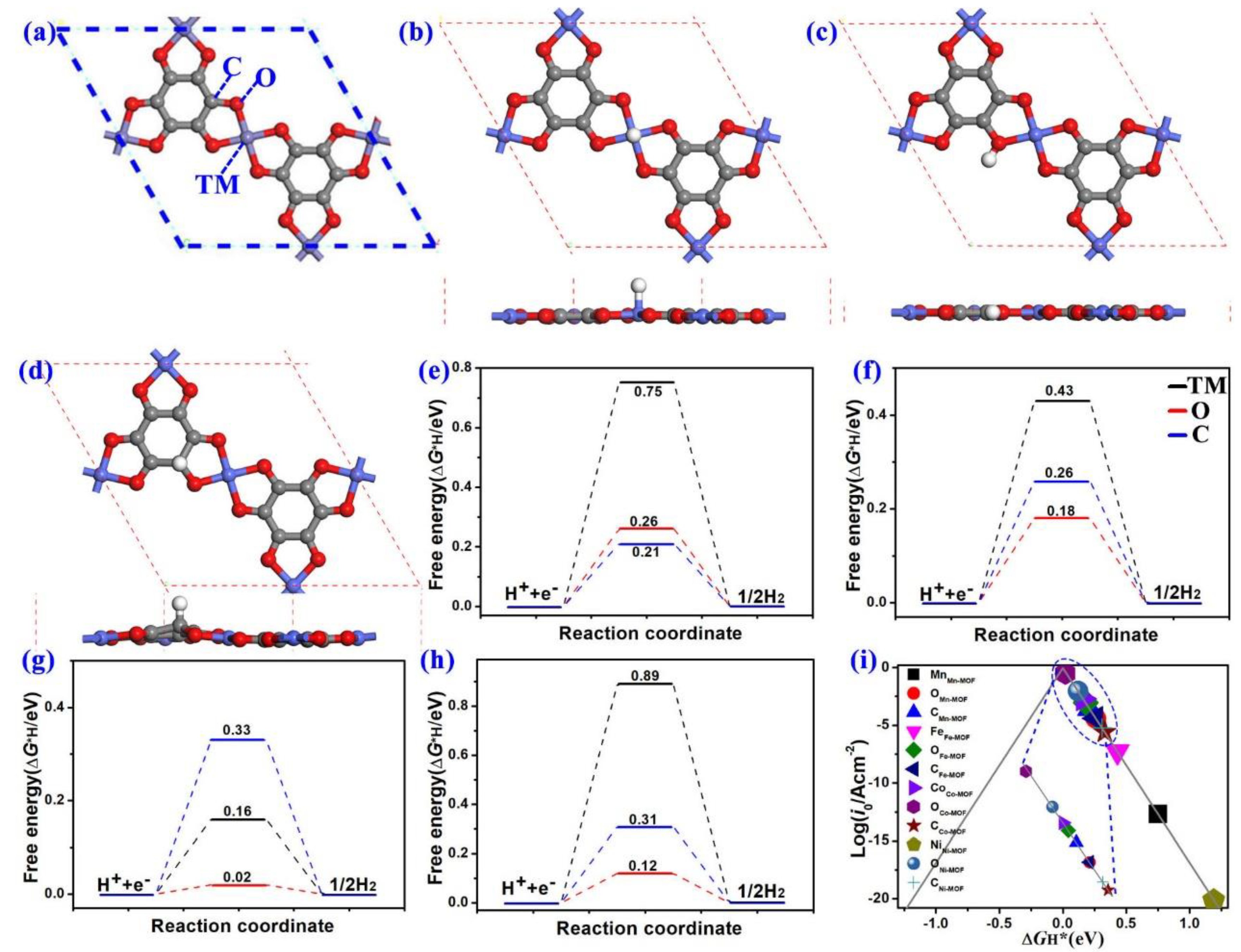
3.3. Hydrogen Evolution Reaction
3.4. Oxygen Evolution Reaction
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Kittner, N.; Lill, F.; Kammen, D.M. Energy storage deployment and innovation for the clean energy transition. Nat. Energy 2017, 2, 17125. [Google Scholar] [CrossRef]
- Yuan, W.; Ma, Y.; Wu, H.; Cheng, L. Single-atom catalysts for CO oxidation, CO2 reduction, and O2 electrochemistry. J. Energy Chem. 2022, 65, 254–279. [Google Scholar] [CrossRef]
- Yang, D.H.; Tao, Y.; Ding, X.; Han, B.H. Porous organic polymers for electrocatalysis. Chem. Soc. Rev. 2022, 51, 761–791. [Google Scholar] [CrossRef] [PubMed]
- Vogt, C.; Weckhuysen, B.M. The concept of active site in heterogeneous catalysis. Nat. Rev. Chem. 2022, 6, 89–111. [Google Scholar] [CrossRef]
- Luo, H.; Yu, P.; Li, G.; Yan, K. Topological quantum materials for energy conversion and storage. Nat. Rev. Phys. 2022, 4, 611–624. [Google Scholar] [CrossRef]
- Zhang, X.; Wu, T.; Yu, C.; Lu, R. Ultrafast Interlayer Charge Separation, Enhanced Visible-Light Absorption, and Tunable Overpotential in Twisted Graphitic Carbon Nitride Bilayers for Water Splitting. Adv. Mater. 2021, 33, e2104695. [Google Scholar] [CrossRef]
- Lui, Y.H.; Zhang, B.; Hu, S. Rational design of photoelectrodes for photoelectrochemical water splitting and CO2 reduction. Front. Phys. 2019, 14, 53402. [Google Scholar] [CrossRef]
- Chen, Z.; Duan, X.; Wei, W.; Wang, S.; Zhang, Z.; Ni, B.-J. Boride-based electrocatalysts: Emerging candidates for water splitting. Nano Res. 2020, 13, 293–314. [Google Scholar] [CrossRef]
- Li, Y.; Li, Y.-L.; Sa, B.; Ahuja, R. Review of two-dimensional materials for photocatalytic water splitting from a theoretical perspective. Catal. Sci. Tech. 2017, 7, 545–559. [Google Scholar] [CrossRef]
- Liu, Y.; Vijayakumar, P.; Liu, Q.; Sakthivel, T.; Chen, F.; Dai, Z. Shining Light on Anion-Mixed Nanocatalysts for Efficient Water Electrolysis: Fundamentals, Progress, and Perspectives. Nanomicro Lett. 2022, 14, 43. [Google Scholar] [CrossRef]
- Su, T.; Shao, Q.; Qin, Z.; Guo, Z.; Wu, Z. Role of Interfaces in Two-Dimensional Photocatalyst for Water Splitting. ACS Catal. 2018, 8, 2253–2276. [Google Scholar] [CrossRef]
- Paul, R.; Zhu, L.; Chen, H.; Qu, J.; Dai, L. Recent Advances in Carbon-Based Metal-Free Electrocatalysts. Adv. Mater. 2019, 31, e1806403. [Google Scholar] [CrossRef]
- Qiao, B.; Wang, A.; Yang, X.; Allard, L.F.; Jiang, Z.; Cui, Y.; Liu, J.; Li, J.; Zhang, T. Single-atom catalysis of CO oxidation using Pt1/FeOx. NatChem 2011, 3, 634–641. [Google Scholar] [CrossRef]
- Xue, Z.-H.; Luan, D.; Zhang, H.; Lou, X.W. Single-atom catalysts for photocatalytic energy conversion. Joule 2022, 6, 92–133. [Google Scholar] [CrossRef]
- Wu, X.; Zhang, H.; Zuo, S.; Dong, J.; Li, Y.; Zhang, J.; Han, Y. Engineering the Coordination Sphere of Isolated Active Sites to Explore the Intrinsic Activity in Single-Atom Catalysts. Nanomicro Lett. 2021, 13, 136. [Google Scholar] [CrossRef]
- Zhou, Y.; Gao, G.; Kang, J.; Chu, W.; Wang, L.-W. Transition metal-embedded two-dimensional C3N as a highly active electrocatalyst for oxygen evolution and reduction reactions. J. Mater. Chem. A 2019, 7, 12050–12059. [Google Scholar] [CrossRef]
- Wang, X.; Zhang, Y.; Wu, J.; Zhang, Z.; Liao, Q.; Kang, Z.; Zhang, Y. Single-Atom Engineering to Ignite 2D Transition Metal Dichalcogenide Based Catalysis: Fundamentals, Progress, and Beyond. Chem. Rev. 2022, 122, 1273–1348. [Google Scholar] [CrossRef]
- Lei, Z.; Sathish, C.I.; Liu, Y.; Karokoti, A.; Wang, J.; Qiao, L.; Vinu, A.; Yi, J. Single metal atoms catalysts—Promising candidates for next generation energy storage and conversion devices. EcoMat 2022, 4, e12186. [Google Scholar] [CrossRef]
- Cao, L.; Luo, Q.; Chen, J.; Wang, L.; Lin, Y.; Wang, H.; Liu, X.; Shen, X.; Zhang, W.; Liu, W.; et al. Dynamic oxygen adsorption on single-atomic Ruthenium catalyst with high performance for acidic oxygen evolution reaction. Nat. Commun. 2019, 10, 4849. [Google Scholar] [CrossRef]
- Yu, M.; Dong, R.; Feng, X. Two-Dimensional Carbon-Rich Conjugated Frameworks for Electrochemical Energy Applications. J. Am. Chem. Soc. 2020, 142, 12903–12915. [Google Scholar] [CrossRef]
- Meng, H.; Han, Y.; Zhou, C.; Jiang, Q.; Shi, X.; Zhan, C.; Zhang, R. Conductive Metal–Organic Frameworks: Design, Synthesis, and Applications. Small Methods 2020, 4, 2000396. [Google Scholar] [CrossRef]
- Lin, L.; Zhang, Q.; Ni, Y.; Shang, L.; Zhang, X.; Yan, Z.; Zhao, Q.; Chen, J. Rational design and synthesis of two-dimensional conjugated metal-organic polymers for electrocatalysis applications. Chem 2022, 8, 1822–1854. [Google Scholar] [CrossRef]
- Ye, Z.; Jiang, Y.; Li, L.; Wu, F.; Chen, R. Rational Design of MOF-Based Materials for Next-Generation Rechargeable Batteries. Nanomicro Lett. 2021, 13, 203. [Google Scholar] [CrossRef]
- Wang, M.; Dong, R.; Feng, X. Two-dimensional conjugated metal-organic frameworks (2D c-MOFs): Chemistry and function for MOF tronics. Chem. Soc. Rev. 2021, 50, 2764–2793. [Google Scholar] [CrossRef]
- Li, C.; Zhang, L.; Chen, J.; Li, X.; Sun, J.; Zhu, J.; Wang, X.; Fu, Y. Recent development and applications of electrical conductive MOFs. Nanoscale 2021, 13, 485–509. [Google Scholar] [CrossRef]
- Dong, R.; Zhang, Z.; Tranca, D.C.; Zhou, S.; Wang, M.; Adler, P.; Liao, Z.; Liu, F.; Sun, Y.; Shi, W.; et al. A coronene-based semiconducting two-dimensional metal-organic framework with ferromagnetic behavior. Nat. Commun. 2018, 9, 2637. [Google Scholar] [CrossRef]
- Miner, E.M.; Fukushima, T.; Sheberla, D.; Sun, L.; Surendranath, Y.; Dinca, M. Electrochemical oxygen reduction catalysed by Ni3(hexaiminotriphenylene)2. Nat. Commun. 2016, 7, 10942. [Google Scholar] [CrossRef]
- Chen, X.; Sun, F.; Bai, F.; Xie, Z. DFT study of the two dimensional metal–organic frameworks X3(HITP)2 as the cathode electrocatalysts for fuel cell. Appl. Surf. Sci. 2019, 471, 256–262. [Google Scholar] [CrossRef]
- Feng, Z.; Li, Y.; Ma, Y.; An, Y.; Dai, X. Magnetic and electronic properties of two-dimensional metal-organic frameworks TM3(C2NH)12. Chin. Phys. B 2021, 30, 097102. [Google Scholar] [CrossRef]
- Yang, X.; Feng, Z.; Guo, Z. Theoretical Investigation on the Hydrogen Evolution, Oxygen Evolution, and Oxygen Reduction Reactions Performances of Two-Dimensional Metal-Organic Frameworks Fe3(C2X)12 (X = NH, O, S). Molecules 2022, 27, 1528. [Google Scholar] [CrossRef]
- Park, J.; Hinckley, A.C.; Huang, Z.; Feng, D.; Yakovenko, A.A.; Lee, M.; Chen, S.; Zou, X.; Bao, Z. Synthetic Routes for a 2D Semiconductive Copper Hexahydroxybenzene Metal-Organic Framework. J. Am. Chem. Soc. 2018, 140, 14533–14537. [Google Scholar] [CrossRef]
- Kresse, G.; Furthmüller, J. Efficient Iterative Schemes for Ab Initio Total-Energy Calculations Using a Plane-Wave Basis Set. Phys. Rev. B 1996, 54, 11169–11186. [Google Scholar] [CrossRef]
- Perdew, J.P.; Chevary, J.A.; Vosko, S.H.; Jackson, K.A.; Pederson, M.R.; Singh, D.J.; Fiolhais, C. Atoms, molecules, solids, and surfaces: Applications of the generalized gradient approximation for exchange and correlation. Phys. Rev. B 1992, 46, 6671–6687. [Google Scholar] [CrossRef]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized gradient approximation made simple. Phys. Rev. Lett. 1996, 77, 3865–3868. [Google Scholar] [CrossRef]
- Grimme, S. Semiempirical gga-type density functional constructed with a longrange dispersion correction. J. Comput. Chem. 2006, 27, 1787–1799. [Google Scholar] [CrossRef]
- Wang, V.; Xu, N.; Liu, J.C.; Tang, G.; Geng, W.T. VASPKIT: A user-friendly Interface Facilitating High-throughput Computing and Analysis Using VASP Code. Comput. Phys. Commun. 2019, 267, 108033. [Google Scholar] [CrossRef]
- Martyna, G.J.; Klein, M.L.; Tuckerman, M. Nosé–hoover chains: The canonical ensemble via continuous dynamics. J. Chem. Phys. 1992, 97, 2635–2643. [Google Scholar] [CrossRef]
- Mathew, K.; Sundararaman, R.; Letchworth-Weaver, K.; Arias, T.A.; Hennig, R.G. Implicit solvation model for density-functional study of nanocrystal surfaces and reaction pathways. J. Chem. Phys. 2014, 140, 084106. [Google Scholar] [CrossRef]
- Nørskov, J.K.; Rossmeisl, J.; Logadottir, A.; Lindqvist, L.; Kitchin, J.R.; Ligaard, T.; Jo’nsson, H. Origin of the Overpotential for Oxygen Reduction at a Fuel-Cell Cathode. J. Phys. Chem. B 2004, 108, 17886–17892. [Google Scholar] [CrossRef]
- Zhang, J.; Zhou, Z.; Wang, F.; Li, Y.; Jing, Y. Two-Dimensional Metal Hexahydroxybenzene Frameworks as Promising Electrocatalysts for an Oxygen Reduction Reaction. ACS Sustain. Chem. Eng. 2020, 8, 7472–7479. [Google Scholar] [CrossRef]
- Chanier, T.; Sargolzaei, M.; Opahle, I.; Hayn, R.; Koepernik, K. LSDA+Uversus LSDA: Towards a better description of the magnetic nearest-neighbor exchange coupling in Co- and Mn-doped ZnO. Phys. Rev. B 2006, 73, 134418. [Google Scholar] [CrossRef]
- Kattel, S.; Atanassov, P.; Kiefer, B. Stability, Electronic and Magnetic Properties of In-Plane Defects in Graphene: A First-Principles Study. J. Phys. Chem. C 2012, 116, 8161–8166. [Google Scholar] [CrossRef]
- Kattel, S.; Atanassov, P.; Kiefer, B. Density Functional Theory Study of Ni–Nx/C Electrocatalyst for Oxygen Reduction in Alkaline and Acidic Media. J. Phys. Chem. C 2012, 116, 17378–17383. [Google Scholar] [CrossRef]
- Yu, L.; Li, F.; Zhao, J.; Chen, Z. Revisiting catalytic performance of supported metal dimers for oxygen reduction reaction via magnetic coupling from first principles. Adv. Powder Mater. 2022, 1, 100031. [Google Scholar] [CrossRef]
- Hu, C.; Song, E.; Wang, M.; Chen, W.; Huang, F.; Feng, Z.; Liu, J.; Wang, J. Partial-Single-Atom, Partial-Nanoparticle Composites Enhance Water Dissociation for Hydrogen Evolution. Adv. Sci. 2021, 8, 2001881. [Google Scholar] [CrossRef]
- Nørskov, J.K.; Bligaard, T.; Logadottir, A.; Kitchin, J.R.; Chen, J.G.; Pandelov, S.; Stimming, U. Trends in the Exchange Current for Hydrogen Evolution. J. ElectroChem. Soc. 2005, 152, 23–26. [Google Scholar] [CrossRef]
- Di Liberto, G.; Cipriano, L.A.; Pacchioni, G. Universal Principles for the Rational Design of Single Atom Electrocatalysts? Handle with Care. ACS Catal. 2022, 12, 5846–5856. [Google Scholar] [CrossRef]
- Talib, S.H.; Lu, Z.; Yu, X.; Ahmad, K.; Bashir, B.; Yang, Z.; Li, J. Theoretical Inspection of M1/PMA Single-Atom Electrocatalyst: Ultra-High Performance for Water Splitting (HER/OER) and Oxygen Reduction Reactions (OER). ACS Catal. 2021, 11, 8929–8941. [Google Scholar] [CrossRef]
- Feng, Z.; Yang, Z.; Meng, X.; Li, F.; Guo, Z.; Zheng, S.; Su, G.; Ma, Y.; Tang, Y.; Dai, X. Two-dimensional metal–organic framework Mo3(C2O)12 as a promising single-atom catalyst for selective nitrogen-to-ammonia conversion. J. Mater. Chem. A 2022, 10, 4731–4738. [Google Scholar] [CrossRef]
- Zhong, W.; Qiu, Y.; Shen, H.; Wang, X.; Yuan, J.; Jia, C.; Bi, S.; Jiang, J. Electronic Spin Moment As a Catalytic Descriptor for Fe Single-Atom Catalysts Supported on C2N. J. Am. Chem. Soc. 2021, 143, 4405–4413. [Google Scholar] [CrossRef]
- Gao, X.; Zhou, Y.; Tan, Y.; Liu, S.; Cheng, Z.; Shen, Z. Graphyne doped with transition-metal single atoms as effective bifunctional electrocatalysts for water splitting. App. Surf. Sci. 2019, 492, 8–15. [Google Scholar] [CrossRef]
- Zhang, W.; Bu, H.; Wang, J.; Zhao, L.; Qu, Y.; Zhao, M. Multi-functional photocatalytic activity of transition-metal tetraaza[14]annulene frameworks. J. Mater. Chem. A 2021, 9, 4221–4229. [Google Scholar] [CrossRef]
- Dang, Q.; Lin, H.; Fan, Z.; Ma, L.; Shao, Q.; Ji, Y.; Zheng, F.; Geng, S.; Yang, S.Z.; Kong, N.; et al. Iridium metallene oxide for acidic oxygen evolution catalysis. Nat. Commun. 2021, 12, 6007. [Google Scholar] [CrossRef]
- Zhang, H.; Wei, W.; Wang, S.; Wang, H.; Huang, B.; Dai, Y. H4,4,4-graphyne with double Dirac points as high-efficiency bifunctional electrocatalysts for water splitting. J. Mate Chem. A 2021, 9, 4082–4090. [Google Scholar] [CrossRef]
- Viswanathan, V.; Hansen, H.A.; Rossmeisl, J.; Nørskov, J.K. Universality in Oxygen Reduction Electrocatalysis on Metal Surfaces. ACS Catal. 2012, 2, 1654–1660. [Google Scholar] [CrossRef]
- Lima, F.H.B.; Zhang, J.; Shao, M.H.; Sasaki, K.; Vukmirovic, M.B.; Ticianelli, E.A.; Adzic, R.R. Catalytic Activity-d-Band Center Correlation for the O2 Reduction Reaction on Platinum in Alkaline Solutions. J. Phys. Chem. C 2007, 111, 404–410. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Materials | a (Å) | DTM-O (Å) | DC-O (Å) | DP (Å) | DTM (Å) | Mtot (μB) | QTM (e) | QO (e) | εd (eV) |
|---|---|---|---|---|---|---|---|---|---|
| Mn-O-MOF | 13.25 | 1.88 | 1.33 | 13.25 | 6.62 | 9.00 | −1.54 | +0.77 | −1.76 |
| Fe-O-MOF | 13.15 | 1.85 | 1.32 | 13.15 | 6.57 | 6.00 | −1.23 | +0.72 | −1.97 |
| Co-O-MOF | 13.04 | 1.84 | 1.31 | 13.04 | 6.52 | 3.73 | −0.99 | +0.71 | −1.88 |
| Ni-O-MOF | 12.98 | 1.83 | 1.30 | 12.98 | 6.49 | 0.50 | −0.88 | +0.69 | −2.09 |
| Materials | ΔGOH* (eV) | ΔGO* (eV) | ΔGOOH* (eV) | ηOER (V) | Uwork (V) | PLS |
|---|---|---|---|---|---|---|
| Mn-O-MOF | 0.91 | 2.14 | 4.01 | 0.64 | 1.87 | *O → *OOH |
| Fe-O-MOF | 0.86 | 2.09 | 4.04 | 0.72 | 1.95 | *O → *OOH |
| Co-O-MOF | 1.40 | 2.72 | 4.48 | 0.53 | 1.76 | *O → *OOH |
| Ni-O-MOF | 2.11 | 3.64 | 5.03 | 0.88 | 2.11 | * → *OH |
| Ideal | 1.23 | 2.46 | 3.69 | 0.00 | 1.23 | - |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, Q.; Feng, Z.; Wang, D. Theoretical Study on the Electrochemical Water Splitting of Two-Dimensional Metal–Organic Frameworks TM3C12O12 (TM = Mn, Fe, Co, Ni). Crystals 2022, 12, 1289. https://doi.org/10.3390/cryst12091289
Li Q, Feng Z, Wang D. Theoretical Study on the Electrochemical Water Splitting of Two-Dimensional Metal–Organic Frameworks TM3C12O12 (TM = Mn, Fe, Co, Ni). Crystals. 2022; 12(9):1289. https://doi.org/10.3390/cryst12091289
Chicago/Turabian StyleLi, Quan, Zhen Feng, and Dianhui Wang. 2022. "Theoretical Study on the Electrochemical Water Splitting of Two-Dimensional Metal–Organic Frameworks TM3C12O12 (TM = Mn, Fe, Co, Ni)" Crystals 12, no. 9: 1289. https://doi.org/10.3390/cryst12091289