
Controlling the Polymorphic Outcome of 2,6-Dimethoxybenzoic Acid Crystallization Using Additives


Abstract
:1. Introduction
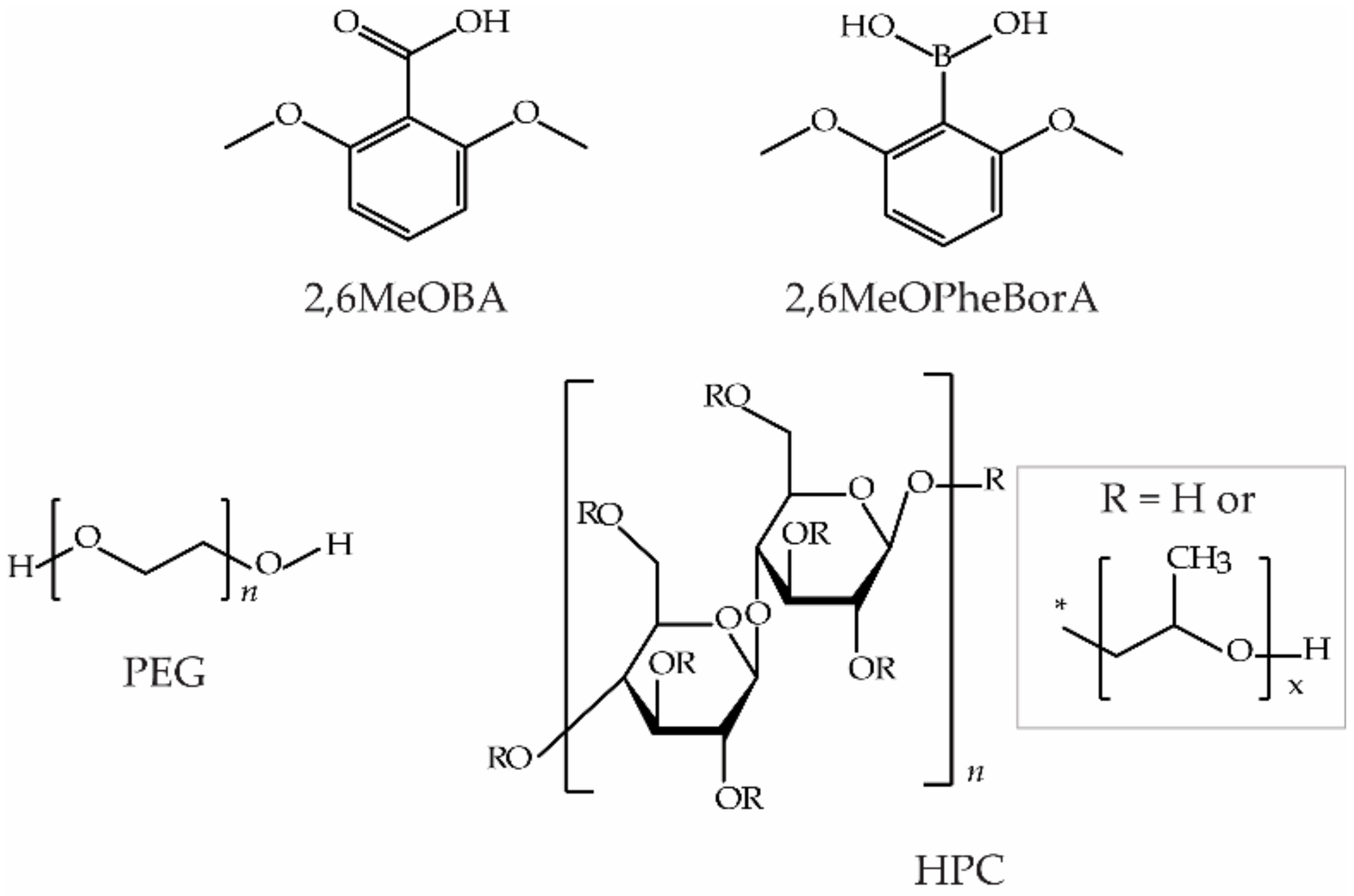
2. Materials and Methods
2.1. Crystallization Experiments
2.2. Solubility Measurements and Solvent-MediatedPhase Transition (SMPT) Studies
2.3. Solid Phase Characterization
2.4. Rietveld Refinement for Form Quantification
3. Results and Discussion
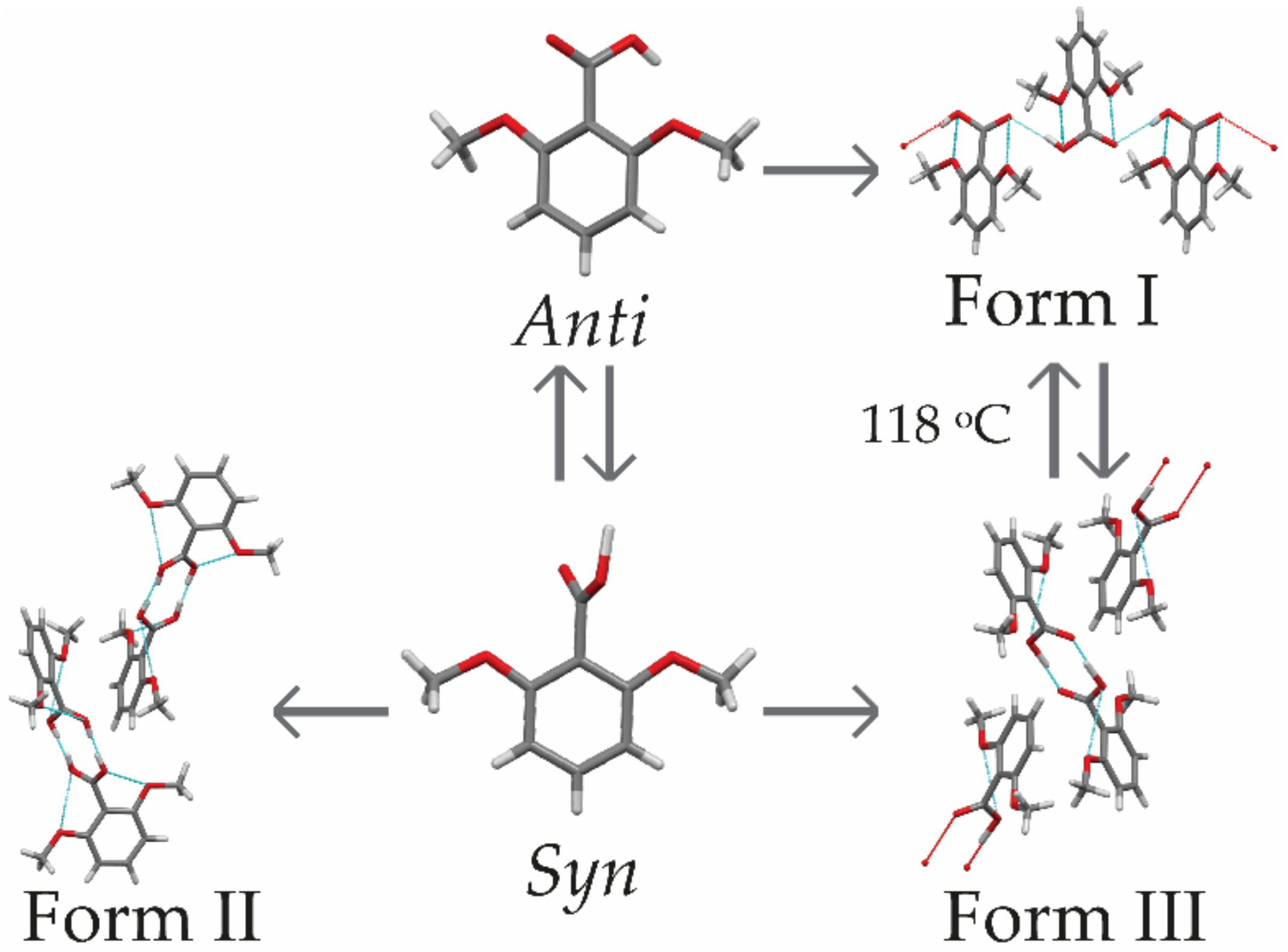
3.1. Characterization of 2,6MeOBA Polymorph
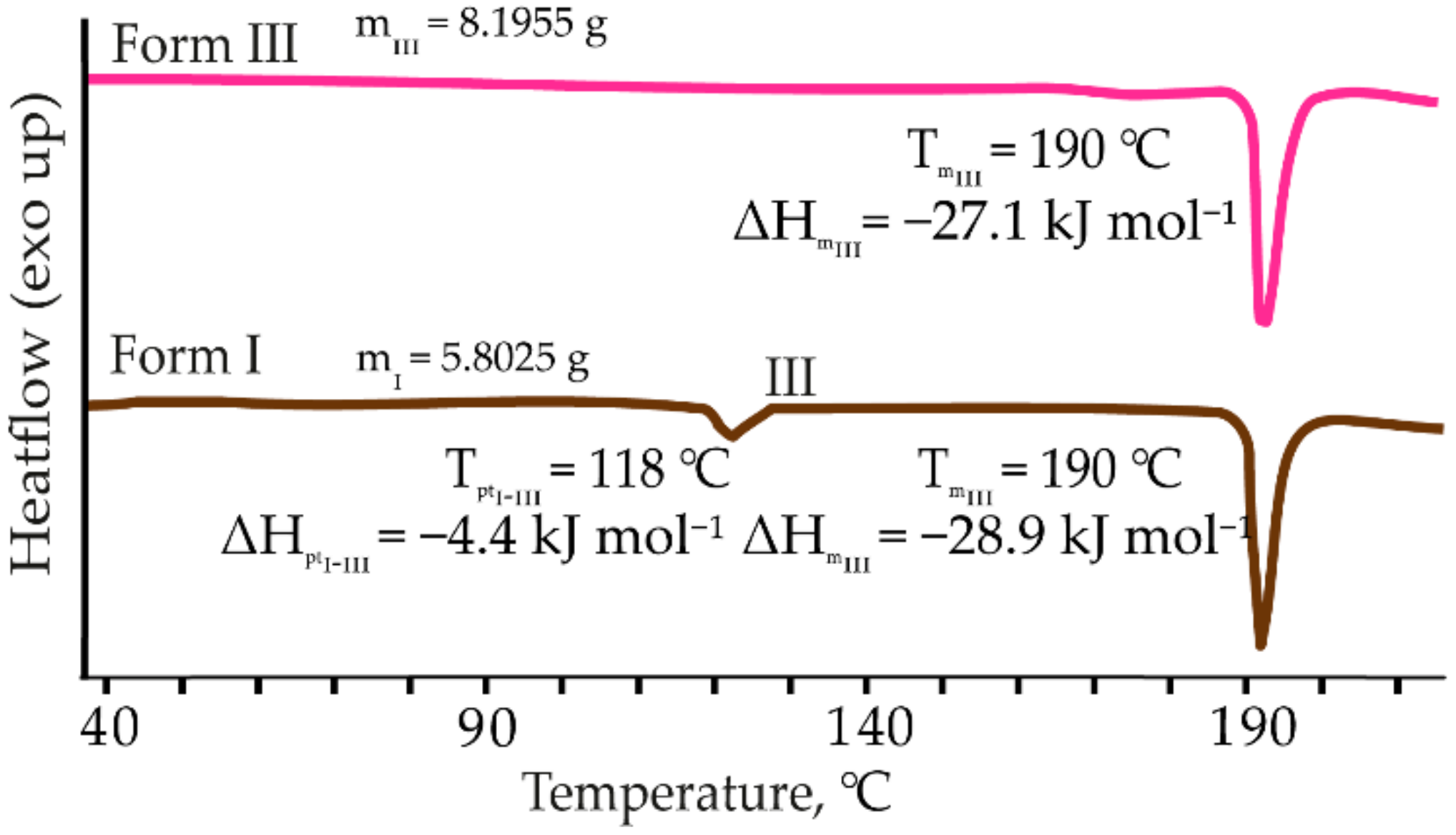
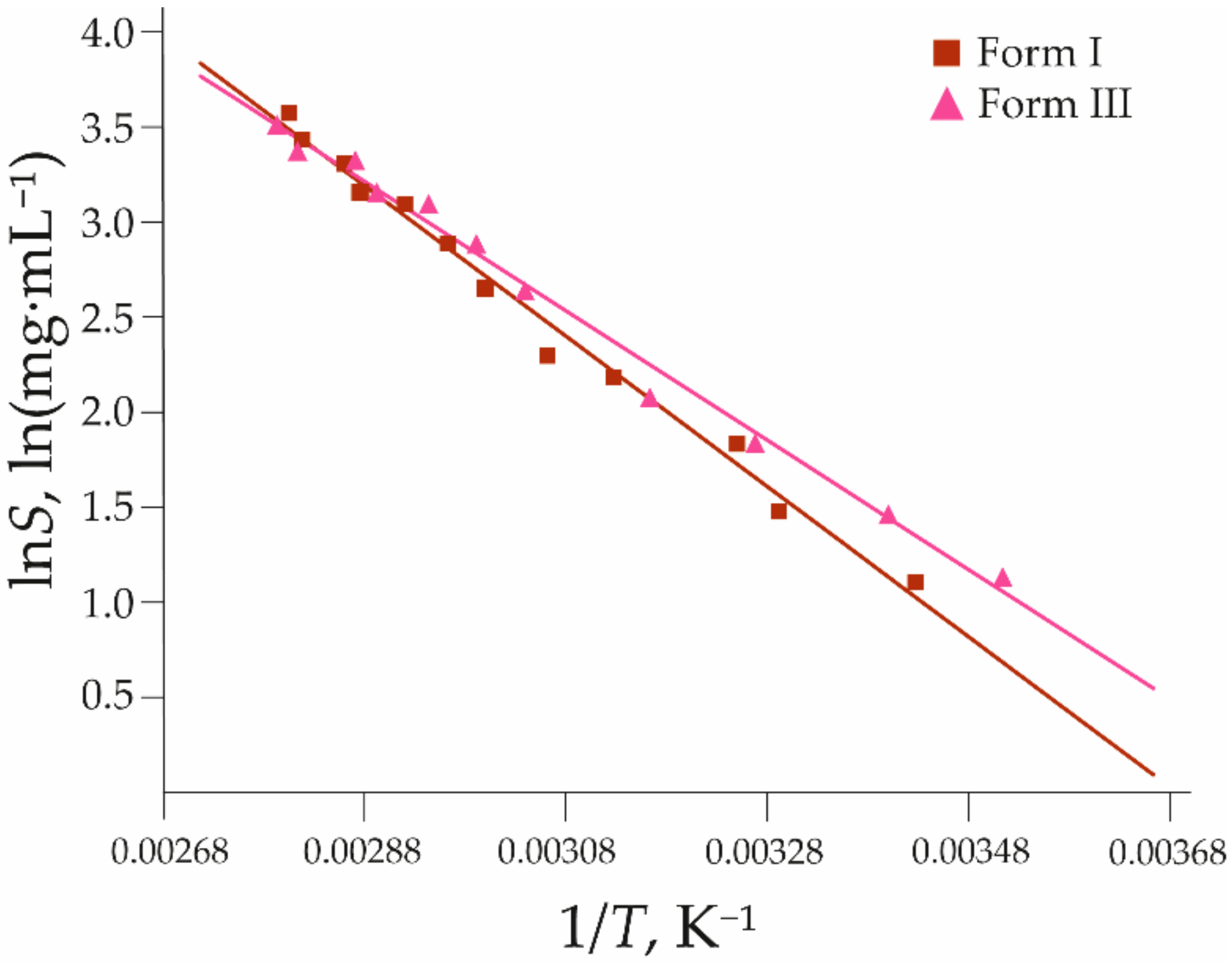
3.1.1. Thermal Characterization and Solubility
3.1.2. Solvent-Mediated Phase Transition (SMPT)
3.2. Crystallization from Pure Solvents
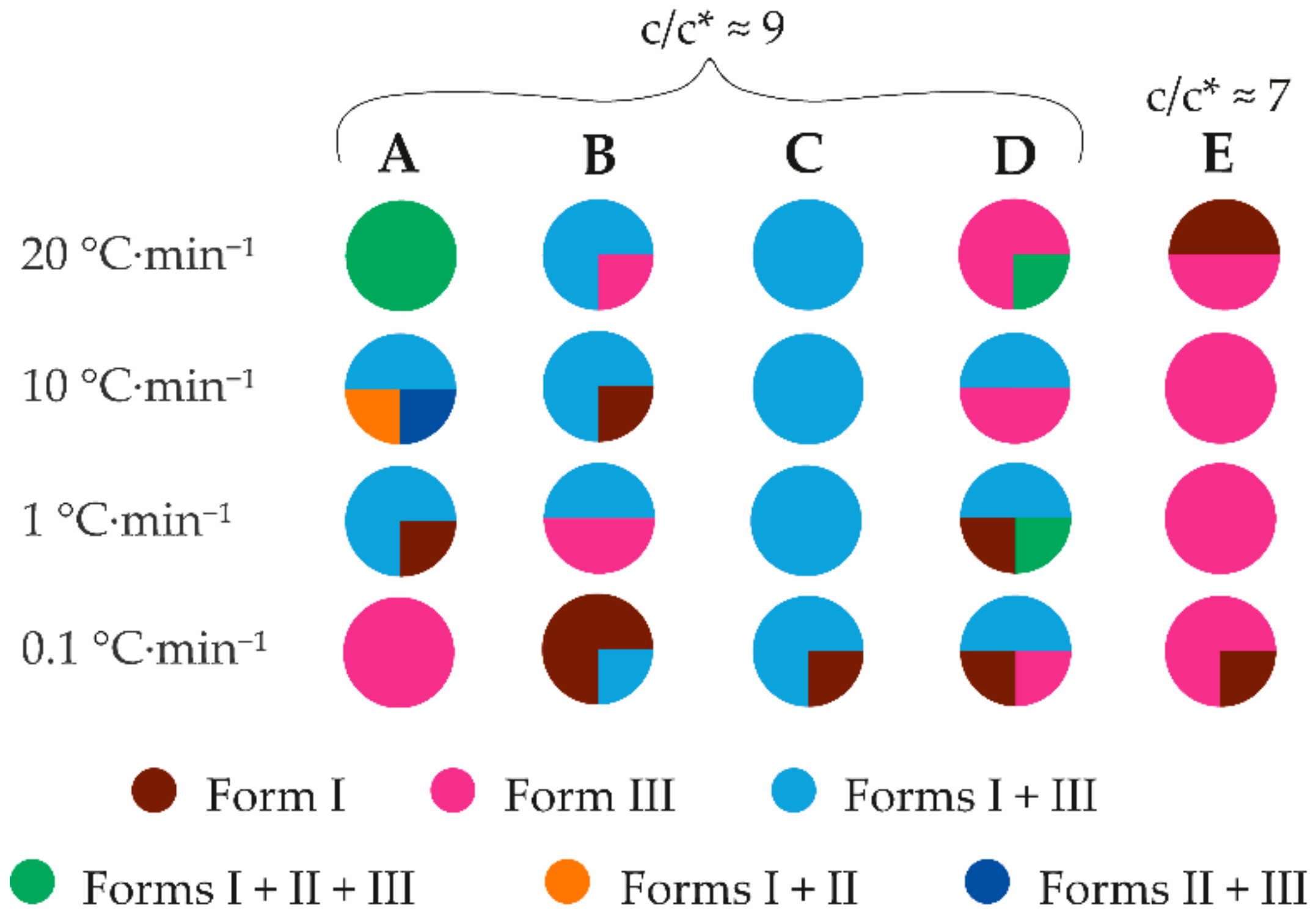
3.3. Crystallization with Additives
3.3.1. Crystallization from Highly Concentrated Water Solution
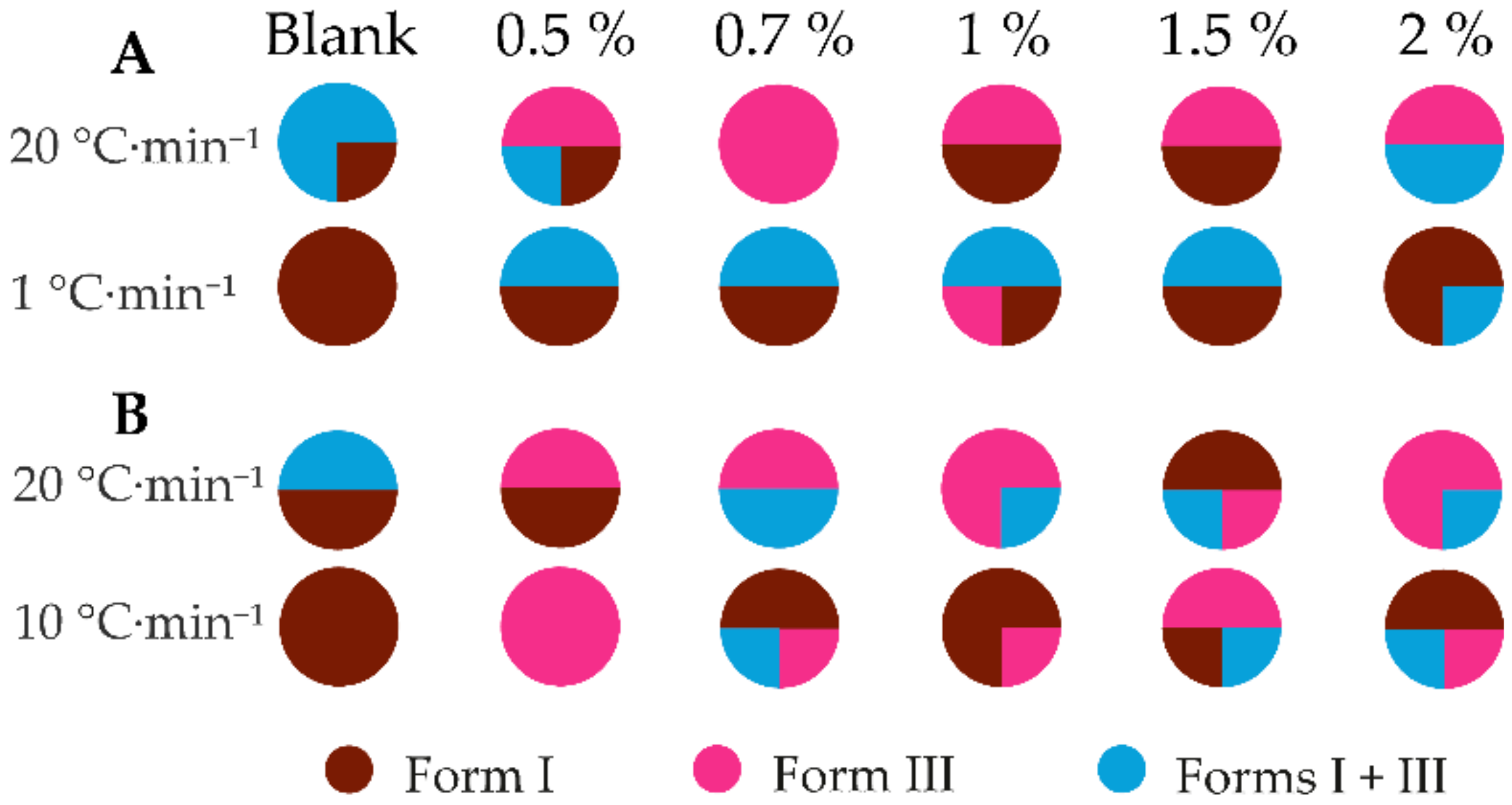
3.3.2. The Effect of the Additive Quantity on the Crystallization Outcome
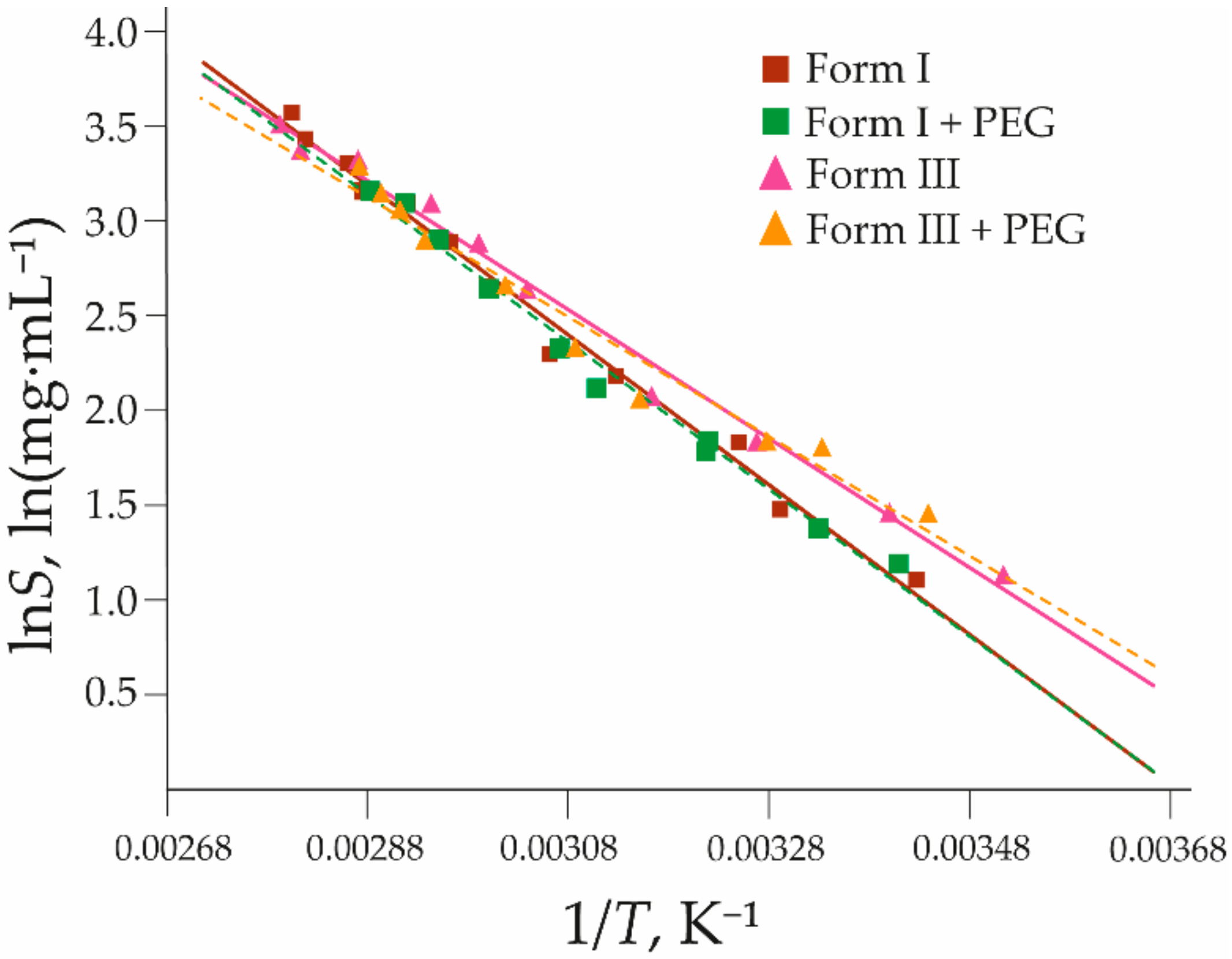
3.4. Effect of Additives on Polymorph Solubility and Solvent-Mediated Phase Transitions
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
References
- Aitipamula, S.; Nangia, A. Polymorphism: Fundamentals and Applications. In Supramolecular Chemistry; John Wiley & Sons, Ltd.: Chichester, UK, 2012; ISBN 9780470661345. [Google Scholar]
- Hilfiker, R. Polymorphism: In the Pharmaceutical Industry; Hilkifer, R., Ed.; Wiley-VCH Verlag GmbH & Co. KGaA: Weinheim, Germany, 2006; ISBN 9783527311460. [Google Scholar]
- Fabbiani, F.P.A.; Allan, D.R.; Parsons, S.; Pulham, C.R. An Exploration of the Polymorphism of Piracetam Using High Pressure. CrystEngComm 2005, 7, 179–186. [Google Scholar] [CrossRef]
- Karpinski, P.H. Polymorphism of Active Pharmaceutical Ingredients. Chem. Eng. Technol. 2006, 29, 233–237. [Google Scholar] [CrossRef]
- Lu, J.; Rohani, S. Polymorphism and Crystallization of Active Pharmaceutical Ingredients (APIs). Curr. Med. Chem. 2009, 16, 884–905. [Google Scholar] [CrossRef]
- Tandon, R.; Tandon, N.; Thapar, R.K. Patenting of Polymorphs. Pharm. Pat. Anal. 2018, 7, 59–63. [Google Scholar] [CrossRef] [PubMed]
- Rakoczy, W.A.; Mazzochi, D.M. The Case of the Disappearing Polymorph: “Inherent Anticipation” and the Impact of SmithKline Beecham Corp. v Apotex Corp. (Paxil®) on Patent Validity and Infringement by Inevitable Conversion. J. Generic Med. 2006, 3, 131–139. [Google Scholar] [CrossRef]
- Snider, D.A.; Addicks, W.; Owens, W. Polymorphism in Generic Drug Product Development. Adv. Drug Deliv. Rev. 2004, 56, 391–395. [Google Scholar] [CrossRef]
- Pudipeddi, M.; Serajuddin, A.T.M. Trends in Solubility of Polymorphs. J. Pharm. Sci. 2005, 94, 929–939. [Google Scholar] [CrossRef] [PubMed]
- De Tros Ilarduya, M.C.; Martín, C.; Goñi, M.M.; Martínez-Uhárriz, M.C. Dissolution Rate of Polymorphs and Two New Pseudopolymorphs of Sulindac. Drug Dev. Ind. Pharm. 1997, 23, 1095–1098. [Google Scholar] [CrossRef]
- Gu, C.H.; Grant, D.J.W. Estimating the Relative Stability of Polymorphs and Hydrates from Heats of Solution and Solubility Data. J. Pharm. Sci. 2001, 90, 1277–1287. [Google Scholar] [CrossRef] [PubMed]
- Vippagunta, S.R.; Brittain, H.G.; Grant, D.J.W. Crystalline Solids. Adv. Drug Deliv. Rev. 2001, 48, 3–26. [Google Scholar] [CrossRef]
- Gupta, S.; Kesarla, R.; Omri, A. Formulation Strategies to Improve the Bioavailability of Poorly Absorbed Drugs with Special Emphasis on Self-Emulsifying Systems. ISRN Pharm. 2013, 2013, 848043. [Google Scholar] [CrossRef] [PubMed]
- Bauer, J.; Spanton, S.; Henry, R.; Quick, J.; Dziki, W.; Porter, W.; Morris, J. Ritonavir: An Extraordinary Example of Conformational Polymorphism. Pharm. Res. 2001, 18, 859–866. [Google Scholar] [CrossRef] [PubMed]
- Aguiar, A.J.; Krc, J.; Kinkel, A.W.; Samyn, J.C. Effect of Polymorphism on the Absorption of Chloramphenicol from Chloramphenicol Palmitate. J. Pharm. Sci. 1967, 56, 847–853. [Google Scholar] [CrossRef] [PubMed]
- Singhal, D.; Curatolo, W. Drug Polymorphism and Dosage Form Design: A Practical Perspective. Adv. Drug Deliv. Rev. 2004, 56, 335–347. [Google Scholar] [CrossRef] [PubMed]
- Censi, R.; Martino, P. di Polymorph Impact on the Bioavailability and Stability of Poorly Soluble Drugs. Molecules 2015, 20, 18759–18776. [Google Scholar] [CrossRef] [PubMed]
- Tang, W.; Sima, A.D.; Gong, J.; Wang, J.; Li, T. Kinetic Difference between Concomitant Polymorphism and Solvent-Mediated Phase Transformation: A Case of Tolfenamic Acid. Cryst. Growth Des. 2020, 20, 1779–1788. [Google Scholar] [CrossRef]
- Su, Y.; Xu, J.; Shi, Q.; Yu, L.; Cai, T. Polymorphism of Griseofulvin: Concomitant Crystallization from the Melt and a Single Crystal Structure of a Metastable Polymorph with Anomalously Large Thermal Expansion. Chem. Commun. 2018, 54, 358–361. [Google Scholar] [CrossRef]
- Du, W.; Yin, Q.; Bao, Y.; Xie, C.; Hou, B.; Hao, H.; Chen, W.; Wang, J.; Gong, J. Concomitant Polymorphism of Prasugrel Hydrochloride in Reactive Crystallization. Ind. Eng. Chem. Res. 2013, 52, 16182–16189. [Google Scholar] [CrossRef]
- Jiang, S.; ter Horst, J.H.; Jansens, P.J. Concomitant Polymorphism of O-Aminobenzoic Acid in Antisolvent Crystallization. Cryst. Growth Des. 2008, 8, 37–43. [Google Scholar] [CrossRef]
- Munshi, P.; Venugopala, K.N.; Jayashree, B.S.; Guru Row, T.N. Concomitant Polymorphism in 3-Acetylcoumarin: Role of Weak C-H⋯O and C-H⋯π Interactions. Cryst. Growth Des. 2004, 4, 1105–1107. [Google Scholar] [CrossRef]
- Singh, A.; Lee, I.S.; Kim, K.; Myerson, A.S. Crystal Growth on Self-Assembled Monolayers. CrystEngComm 2011, 13, 24–32. [Google Scholar] [CrossRef]
- Hernández Espinell, J.R.; López-Mejías, V.; Stelzer, T. Revealing Polymorphic Phase Transformations in Polymer-Based Hot Melt Extrusion Processes. Cryst. Growth Des. 2018, 18, 1995–2002. [Google Scholar] [CrossRef] [PubMed]
- Svard, M.; Rasmuson, Å.C. M-Hydroxybenzoic Acid: Quantifying Thermodynamic Stability and Influence of Solvent on the Nucleation of a Polymorphic System. Cryst. Growth Des. 2013, 13, 1140–1152. [Google Scholar] [CrossRef]
- Telford, R.; Seaton, C.C.; Clout, A.; Buanz, A.; Gaisford, S.; Williams, G.R.; Prior, T.J.; Okoye, C.H.; Munshi, T.; Scowen, I.J. Stabilisation of Metastable Polymorphs: The Case of Paracetamol Form III. Chem. Commun. 2016, 52, 12028–12031. [Google Scholar] [CrossRef] [PubMed]
- Lee, E.H.; Byrn, S.R. Stabilization of Metastable Flufenamic Acid by Inclusion of Mefenamic Acid: Solid Solution or Epilayer? J. Pharm. Sci. 2010, 99, 4013–4022. [Google Scholar] [CrossRef] [PubMed]
- Moshe, H.; Levi, G.; Mastai, Y. Polymorphism Stabilization by Crystal Adsorption on a Self-Assembled Monolayer. CrystEngComm 2013, 15, 9203–9209. [Google Scholar] [CrossRef]
- Simone, E.; Steele, G.; Nagy, Z.K. Tailoring Crystal Shape and Polymorphism Using Combinations of Solvents and a Structurally Related Additive. CrystEngComm 2015, 17, 9370–9379. [Google Scholar] [CrossRef]
- Song, R.Q.; Cölfen, H. Additive Controlled Crystallization. CrystEngComm 2011, 13, 1249–1276. [Google Scholar] [CrossRef]
- Tulli, L.G.; Moridi, N.; Wang, W.; Helttunen, K.; Neuburger, M.; Vaknin, D.; Meier, W.; Shahgaldian, P. Polymorphism Control of an Active Pharmaceutical Ingredient beneath Calixarene-Based Langmuir Monolayers. Chem. Commun. 2014, 50, 3938–3940. [Google Scholar] [CrossRef] [PubMed]
- Moridi, N.; Danylyuk, O.; Suwinska, K.; Shahgaldian, P. Monolayers of an Amphiphilic Para-Carboxy-Calix[4]Arene Act as Templates for the Crystallization of Acetaminophen. J. Colloid Interface Sci. 2012, 377, 450–455. [Google Scholar] [CrossRef] [PubMed]
- Hiremath, R.; Basile, J.A.; Varney, S.W.; Swift, J.A. Controlling Molecular Crystal Polymorphism with Self-Assembled Monolayer Templates. J. Am. Chem. Soc. 2005, 127, 18321–18327. [Google Scholar] [CrossRef]
- Hiremath, R.; Varney, S.W.; Swift, J.A. Selective Growth of a Less Stable Polymorph of 2-Iodo-4-Nitroaniline on a Self-Assembled Monolayer Template. Chem. Commun. 2004, 23, 2676–2677. [Google Scholar] [CrossRef] [PubMed]
- Zhang, B.; Hou, X.; Dang, L.; Wei, H. Selective Polymorphic Crystal Growth on Self-Assembled Monolayer Using Molecular Modeling as an Assistant Method. J. Cryst. Growth 2019, 518, 81–88. [Google Scholar] [CrossRef]
- López-Mejías, V.; Kampf, J.W.; Matzger, A.J. Nonamorphism in Flufenamic Acid and a New Record for a Polymorphic Compound with Solved Structures. J. Am. Chem. Soc. 2012, 134, 9872–9875. [Google Scholar] [CrossRef]
- Simone, E.; Cenzato, M.V.; Nagy, Z.K. A Study on the Effect of the Polymeric Additive HPMC on Morphology and Polymorphism of Ortho-Aminobenzoic Acid Crystals. J. Cryst. Growth 2016, 446, 50–59. [Google Scholar] [CrossRef]
- Caridi, A.; Kulkarni, S.A.; di Profio, G.; Curcio, E.; ter Horst, J.H. Template-Induced Nucleation of Isonicotinamide Polymorphs. Cryst. Growth Des. 2014, 14, 1135–1141. [Google Scholar] [CrossRef]
- Watson, S.; Nie, M.; Wang, L.; Stokes, K. Challenges and Developments of Self-Assembled Monolayers and Polymer Brushes as a Green Lubrication Solution for Tribological Applications. RSC Adv. 2015, 5, 89698–89730. [Google Scholar] [CrossRef]
- Rossi, A.; Savioli, A.; Bini, M.; Capsoni, D.; Massarotti, V.; Bettini, R.; Gazzaniga, A.; Sangalli, M.E.; Giordano, F. Solid-State Characterization of Paracetamol Metastable Polymorphs Formed in Binary Mixtures with Hydroxypropylmethylcellulose. Thermochim. Acta 2003, 406, 55–67. [Google Scholar] [CrossRef]
- Parambil, J.V.; Poornachary, S.K.; Heng, J.Y.Y.; Tan, R.B.H. Template-Induced Nucleation for Controlling Crystal Polymorphism: From Molecular Mechanisms to Applications in Pharmaceutical Processing. CrystEngComm 2019, 21, 4122–4135. [Google Scholar] [CrossRef]
- Bryan, R.F.; White, D.H. 2,6-Dimethoxybenzoic Acid. Acta Crystallogr. Sect. B Struct. Crystallogr. Cryst. Chem. 1982, 38, 1014–1016. [Google Scholar] [CrossRef]
- Portalone, G. Redetermination of 2,6-Dimethoxy-Benzoic Acid. Acta Crystallogr. Sect. E Struct. Rep. Online 2009, 65, o327–o328. [Google Scholar] [CrossRef] [PubMed]
- Portalone, G. A New Polymorph of 2,6-Dimethoxybenzoic Acid. Acta Crystallogr. Sect. E Struct. Rep. Online 2011, 67, o3394–o3395. [Google Scholar] [CrossRef] [PubMed]
- Portalone, G. Crystal Structure and Hirshfeld Surface Analysis of a Third Polymorph of 2,6-Dimethoxybenzoic Acid. Acta Crystallogr. Sect. E Crystallogr. Commun. 2020, 76, 1823–1826. [Google Scholar] [CrossRef] [PubMed]
- Pal, R.; Jelsch, C.; Malaspina, L.A.; Edwards, A.J.; Murshed, M.M.; Grabowsky, S. Syn and Anti Polymorphs of 2,6-Dimethoxy Benzoic Acid and Its Molecular and Ionic Cocrystals: Structural Analysis and Energetic Perspective. J. Mol. Struct. 2020, 1221, 128721. [Google Scholar] [CrossRef]
- Bērziņš, A.; Semjonova, A.; Actiņš, A.; Salvalaglio, M. Speciation of Substituted Benzoic Acids in Solution: Evaluation of Spectroscopic and Computational Methods for the Identification of Associates and Their Role in Crystallization. Cryst. Growth Des. 2021, 21, 4823–4836. [Google Scholar] [CrossRef]
- Brady, J.; Drig, T.; Lee, P.I.; Li, J.X. Polymer Properties and Characterization. In Developing Solid Oral Dosage Forms: Pharmaceutical Theory and Practice, 2nd ed.; Qiu, Y., Chen, Y., Zhang, G., Yu, L., Mantri, R.V., Eds.; Elsevier: Amsterdam, The Netherlands, 2017; pp. 181–223. ISBN 9780128024478. [Google Scholar]
- Reus, M.A.; van der Heijden, A.E.D.M.; ter Horst, J.H. Solubility Determination from Clear Points upon Solvent Addition. Org. Process Res. Dev. 2015, 19, 1004–1011. [Google Scholar] [CrossRef]
- Doebelin, N.; Kleeberg, R. Profex: A Graphical User Interface for the Rietveld Refinement Program BGMN. J. Appl. Crystallogr. 2015, 48, 1573–1580. [Google Scholar] [CrossRef]
- Burger, A.; Ramberger, R. On the Polymorphism of Pharmaceuticals and Other Molecular Crystals. I. Mikrochim. Acta 1979, 72, 259–271. [Google Scholar] [CrossRef]
- Ostwald, W. Studien Über Die Bildung Und Umwandlung Fester Körper. Z. Phys. Chem. 1897, 22U, 289–330. [Google Scholar] [CrossRef]
- Datta, S.; Grant, D.J.W. Effect of Supersaturation on the Crystallization of Phenylbutazone Polymorphs. Cryst. Res. Technol. 2005, 40, 233–242. [Google Scholar] [CrossRef]
- Liu, Y.; van den Berg, M.H.; Alexander, A.J. Supersaturation Dependence of Glycine Polymorphism Using Laser-Induced Nucleation, Sonocrystallization and Nucleation by Mechanical Shock. Phys. Chem. Chem. Phys. 2017, 19, 19386–19392. [Google Scholar] [CrossRef] [PubMed]
- Lin, J.; Shi, P.; Wang, Y.; Wang, L.; Ma, Y.; Liu, F.; Wu, S.; Gong, J. Template Design Based on Molecular and Crystal Structure Similarity to Regulate Conformational Polymorphism Nucleation: The Case of α,ω-Alkanedicarboxylic Acids. IUCrJ 2021, 8, 814–822. [Google Scholar] [CrossRef] [PubMed]
- Wu, H.; Wang, J.; Liu, Q.; Zong, S.; Tian, B.; Huang, X.; Wang, T.; Yin, Q.; Hao, H. Influences and the Mechanism of Additives on Intensifying Nucleation and Growth of P-Methylacetanilide. Cryst. Growth Des. 2020, 20, 973–983. [Google Scholar] [CrossRef]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Polymorph | a | b | R2 | SE for lnScalc |
|---|---|---|---|---|
| I | −3960 ± 130 | 15 ± 0.4 | 0.9894 | 0.09 |
| III | −3400 ± 110 | 13 ± 0.3 | 0.9909 | 0.08 |
| Polymorph | a | b | R2 | SE for lnScalc |
|---|---|---|---|---|
| I + PEG | −3910 ± 150 | 14 ± 0.5 | 0.9885 | 0.08 |
| III + PEG | −3180 ± 200 | 12 ± 0.6 | 0.9698 | 0.12 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Semjonova, A.; Bērziņš, A. Controlling the Polymorphic Outcome of 2,6-Dimethoxybenzoic Acid Crystallization Using Additives. Crystals 2022, 12, 1161. https://doi.org/10.3390/cryst12081161
Semjonova A, Bērziņš A. Controlling the Polymorphic Outcome of 2,6-Dimethoxybenzoic Acid Crystallization Using Additives. Crystals. 2022; 12(8):1161. https://doi.org/10.3390/cryst12081161
Chicago/Turabian StyleSemjonova, Aina, and Agris Bērziņš. 2022. "Controlling the Polymorphic Outcome of 2,6-Dimethoxybenzoic Acid Crystallization Using Additives" Crystals 12, no. 8: 1161. https://doi.org/10.3390/cryst12081161