
A Solid-Solid Phase Transformation of Triclabendazole at High Pressures

 ,
,
Abstract
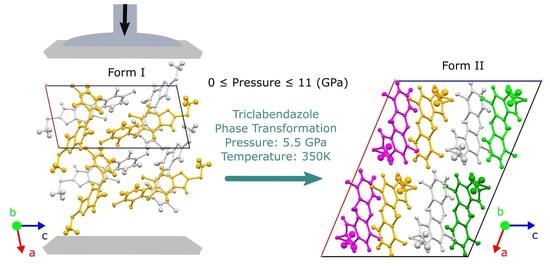
:
1. Introduction
2. Methods
2.1. Structure Optimization and Energy Calculation
2.2. Computation of Raman Spectra
3. Results and Discussion
3.1. Pressure Dependence of Volume
3.2. Gibbs Free Energy Difference
3.3. Vibrational Spectra
3.3.1. FT-IR Spectra
3.3.2. Raman Spectra Calculation
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Mas-Coma, S.; Valero, M.A.; Bargues, M.D. Chapter 2 Fasciola, Lymnaeids and Human Fascioliasis, with a Global Overview on Disease Transmission, Epidemiology, Evolutionary Genetics, Molecular Epidemiology and Control. In Advances in Parasitology; Elsevier: Amsterdam, The Netherlands, 2009; Volume 69, pp. 41–146. ISBN 978-0-12-374795-2. [Google Scholar]
- Ashrafi, K.; Bargues, M.D.; O’Neill, S.; Mas-Coma, S. Fascioliasis: A Worldwide Parasitic Disease of Importance in Travel Medicine. Travel Med. Infect. Dis. 2014, 12, 636–649. [Google Scholar] [CrossRef] [PubMed]
- Mas-Coma, S.; Valero, M.A.; Bargues, M.D. Fascioliasis. In Digenetic Trematodes; Toledo, R., Fried, B., Eds.; Advances in Experimental Medicine and Biology Book Series; Springer International Publishing: Cham, Switzerland, 2019; Volume 1154, pp. 71–103. ISBN 978-3-030-18615-9. [Google Scholar]
- Duthaler, U.; Smith, T.A.; Keiser, J. In Vivo and In Vitro Sensitivity of Fasciola hepatica to Triclabendazole Combined with Artesunate, Artemether, or OZ78. Antimicrob Agents Chemother 2010, 54, 4596–4604. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mas-Coma, S.; Bargues, M.D.; Valero, M.A. Human Fascioliasis Infection Sources, Their Diversity, Incidence Factors, Analytical Methods and Prevention Measures. Parasitology 2018, 145, 1665–1699. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takeba, K.; Fujinuma, K.; Sakamoto, M.; Miyazaki, T.; Oka, H.; Itoh, Y.; Nakazawa, H. Simultaneous Determination of Triclabendazole and Its Sulphoxide and Sulphone Metabolites in Bovine Milk by High-Performance Liquid Chromatography. J. Chromatogr. A 2000, 882, 99–107. [Google Scholar] [CrossRef]
- Fairweather, I. Triclabendazole: New Skills to Unravel an Old(Ish) Enigma. J. Helminthol. 2005, 79, 227–234. [Google Scholar] [CrossRef]
- Gandhi, P.; Schmitt, E.K.; Chen, C.-W.; Samantray, S.; Venishetty, V.K.; Hughes, D. Triclabendazole in the Treatment of Human Fascioliasis: A Review. Trans. R. Soc. Trop. Med. Hyg. 2019, 113, 797–804. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- US Food and Drug Administration. Drug Trials Snapshots: EGATENS Food and Drug Administration. Drug Trials Snapshots: EGATEN. 2019. Available online: https://www.fda.gov/drugs/drug-trials-snapshots-egaten (accessed on 29 April 2019).
- Gallay, J.-J.; Kuhne, M.; Meyer, A.; Rechsteiner, O.; Schellenbaum, M. 2-Alkylthio-, 2-Alkylsulphinyl- and 2-Alkylsulfonyl-6-Phenylbenzimidazoles as Anthelmintic Agents. U.S. Patent 4,197,307, 10 April 1978. [Google Scholar]
- Kuhnert-Brandst, M. Contribution to polymorphism of drugs, 7 communication: Famotidine, flupirtine maleate, GYKI-51189,paxamate, propentofylline and triclabendazole. Sci. Pharm. 1990, 58, 55–67. [Google Scholar]
- Tothadi, S.; Bhogala, B.R.; Gorantla, A.R.; Thakur, T.S.; Jetti, R.K.R.; Desiraju, G.R. Triclabendazole: An Intriguing Case of Co-Existence of Conformational and Tautomeric Polymorphism. Chem. Asian J. 2012, 7, 330–342. [Google Scholar] [CrossRef] [PubMed]
- Salazar-Rojas, D.; Kaufman, T.S.; Maggio, R.M. A Comprehensive Approach toward Concomitant Triclabendazole Polymorphism in Pharmaceutical Products. J. Drug Deliv. Sci. Technol. 2021, 62, 102386. [Google Scholar] [CrossRef]
- Li, J.; Sode, O.; Voth, G.A.; Hirata, S. A Solid–Solid Phase Transition in Carbon Dioxide at High Pressures and Intermediate Temperatures. Nat. Commun. 2013, 4, 2647. [Google Scholar] [CrossRef]
- Tang, J.; Han, Y.; Ali, I.; Luo, H.; Nowak, A.; Li, J. Stability and Phase Transition Investigation of Olanzapine Polymorphs. Chem. Phys. Lett. 2021, 767, 138384. [Google Scholar] [CrossRef]
- He, X.; Zhu, T.; Wang, X.; Liu, J.; Zhang, J.Z.H. Fragment Quantum Mechanical Calculation of Proteins and Its Applications. Acc. Chem. Res. 2014, 47, 2748–2757. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Qi, L.-W.; Zhang, J.Z.H.; He, X. Fragment Quantum Mechanical Method for Large-Sized Ion–Water Clusters. J. Chem. Theory Comput. 2017, 13, 2021–2034. [Google Scholar] [CrossRef] [PubMed]
- Han, Y.; Ali, I.; Wang, Z.; Cai, J.; Wu, S.; Tang, J.; Zhang, L.; Ren, J.; Xiao, R.; Lu, Q.; et al. Machine Learning Accelerates Quantum Mechanics Predictions of Molecular Crystals. Phys. Rep. 2021, 934, 1–71. [Google Scholar] [CrossRef]
- Hao, X.; Liu, J.; Ali, I.; Luo, H.; Han, Y.; Hu, W.; Liu, J.; He, X.; Li, J. Ab Initio Determination of Crystal Stability of Di-p-Tolyl Disulfide. Sci. Rep. 2021, 11, 7076. [Google Scholar] [CrossRef]
- Hao, X.; Liu, J.; Luo, H.; Han, Y.; Hu, W.; Liu, J.; Li, J.; He, X. Crystal Structure Optimization and Gibbs Free Energy Comparison of Five Sulfathiazole Polymorphs by the Embedded Fragment QM Method at the DFT Level. Crystals 2019, 9, 256. [Google Scholar] [CrossRef] [Green Version]
- Luo, H.; Hao, X.; Gong, Y.; Zhou, J.; He, X.; Li, J. Rational Crystal Polymorph Design of Olanzapine. Cryst. Growth Des. 2019, 19, 2388–2395. [Google Scholar] [CrossRef]
- Luo; Liu; He; Li Low-Temperature Polymorphic Transformation of β-Lactam Antibiotics. Crystals 2019, 9, 460. [CrossRef] [Green Version]
- Dennis, J.E., Jr.; Moré, J.J. Quasi-Newton Methods, Motivation and Theory. SIAM Rev. 1977, 19, 46–89. [Google Scholar] [CrossRef] [Green Version]
- Beran, G.J.O. Modeling Polymorphic Molecular Crystals with Electronic Structure Theory. Chem. Rev. 2016, 116, 5567–5613. [Google Scholar] [CrossRef]
- Head, J.D.; Zerner, M.C. A Broyden—Fletcher—Goldfarb—Shanno Optimization Procedure for Molecular Geometries. Chem. Phys. Lett. 1985, 122, 264–270. [Google Scholar] [CrossRef]
- Liu, J.; He, X. Accurate Prediction of Energetic Properties of Ionic Liquid Clusters Using a Fragment-Based Quantum Mechanical Method. Phys. Chem. Chem. Phys. 2017, 19, 20657–20666. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; He, X.; Zhang, J.Z.H.; Qi, L.-W. Hydrogen-Bond Structure Dynamics in Bulk Water: Insights from Ab Initio Simulations with Coupled Cluster Theory. Chem. Sci. 2018, 9, 2065–2073. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 09 Citation; Gaussian, Inc.: Wallingford, CT, USA, 2009. [Google Scholar]
- Han, Y.; Liu, J.; Li, J. Molecular Structure Determination of Solid Carbon Dioxide Phase IV at High Pressures and Temperatures Based on Møller-Plesset Perturbation Theory. Int. J. Quantum Chem. 2020, 120, e26397. [Google Scholar] [CrossRef]
- Ramírez, F.J.; Tuñón, I.; Collado, J.A.; Silla, E. Structural and Vibrational Study of the Tautomerism of Histamine Free-Base in Solution. J. Am. Chem. Soc. 2003, 125, 2328–2340. [Google Scholar] [CrossRef]
- Manimunda, P.; Syed Asif, S.A.; Mishra, M.K. Probing Stress Induced Phase Transformation in Aspirin Polymorphs Using Raman Spectroscopy Enabled Nanoindentation. Chem. Commun. 2019, 55, 9200–9203. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ali, I.; Tang, J.; Han, Y.; Wei, Z.; Zhang, Y.; Li, J. A Solid-Solid Phase Transformation of Triclabendazole at High Pressures. Crystals 2022, 12, 300. https://doi.org/10.3390/cryst12020300
Ali I, Tang J, Han Y, Wei Z, Zhang Y, Li J. A Solid-Solid Phase Transformation of Triclabendazole at High Pressures. Crystals. 2022; 12(2):300. https://doi.org/10.3390/cryst12020300
Chicago/Turabian StyleAli, Imran, Jiequn Tang, Yanqiang Han, Zhiyun Wei, Yongli Zhang, and Jinjin Li. 2022. "A Solid-Solid Phase Transformation of Triclabendazole at High Pressures" Crystals 12, no. 2: 300. https://doi.org/10.3390/cryst12020300